- NF-κB
-
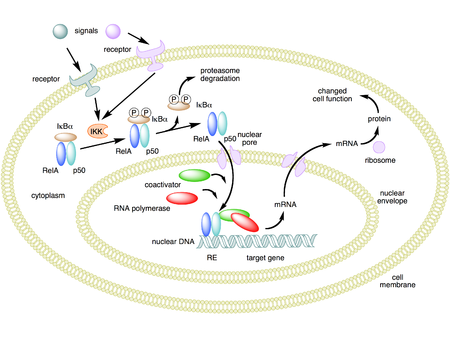 Mechanism of NF-κB action. In this figure, the NF-κB heterodimer between Rel and p50 proteins is used as an example. While in an inactivated state, NF-κB is located in the cytosol complexed with the inhibitory protein IκBα. Through the intermediacy of integral membrane receptors, a variety of extracellular signals can activate the enzyme IκB kinase (IKK). IKK, in turn, phosphorylates the IκBα protein, which results in ubiquitination, dissociation of IκBα from NF-κB, and eventual degradation of IκBα by the proteosome. The activated NF-κB is then translocated into the nucleus where it binds to specific sequences of DNA called response elements (RE). The DNA/NF-κB complex then recruits other proteins such as coactivators and RNA polymerase, which transcribe downstream DNA into mRNA, which, in turn, is translated into protein, which results in a change of cell function.[1][2][3]
Mechanism of NF-κB action. In this figure, the NF-κB heterodimer between Rel and p50 proteins is used as an example. While in an inactivated state, NF-κB is located in the cytosol complexed with the inhibitory protein IκBα. Through the intermediacy of integral membrane receptors, a variety of extracellular signals can activate the enzyme IκB kinase (IKK). IKK, in turn, phosphorylates the IκBα protein, which results in ubiquitination, dissociation of IκBα from NF-κB, and eventual degradation of IκBα by the proteosome. The activated NF-κB is then translocated into the nucleus where it binds to specific sequences of DNA called response elements (RE). The DNA/NF-κB complex then recruits other proteins such as coactivators and RNA polymerase, which transcribe downstream DNA into mRNA, which, in turn, is translated into protein, which results in a change of cell function.[1][2][3]
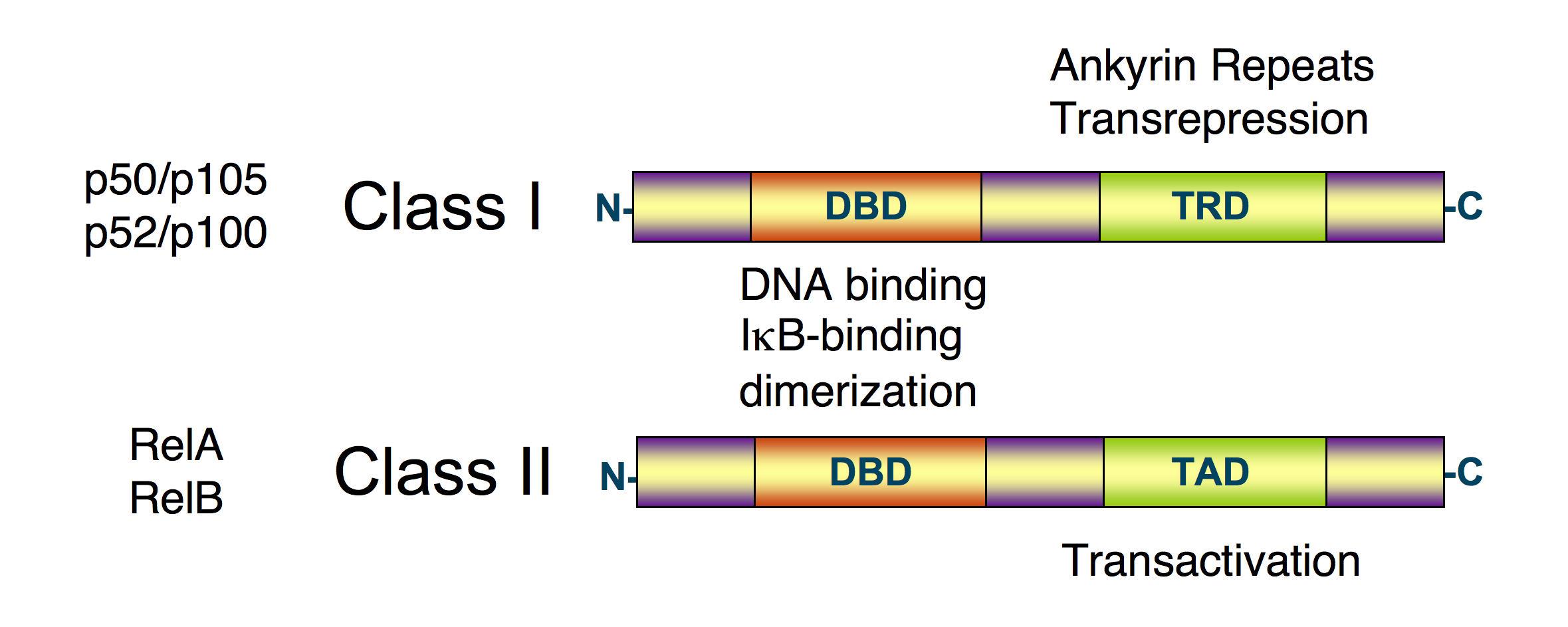
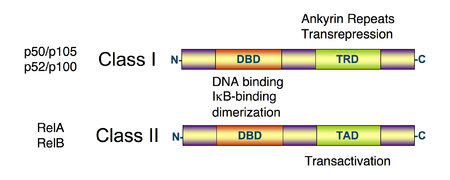 Schematic diagram of NF-κB protein structure. There are two structural classes of NF-κB proteins: class I (top) and class II (bottom). Both classes of proteins contain a N-terminal DNA-binding domain (DBD), which also serves as a dimerization interface to other NF-κB transcription factors and, in addition, binds to the inhibitory IκBα protein. The C-terminus of class I proteins contains a number of ankyrin repeats and has transrepression activity. In contrast, the C-terminus of class II proteins has a transactivation function.[1][2][3]
Schematic diagram of NF-κB protein structure. There are two structural classes of NF-κB proteins: class I (top) and class II (bottom). Both classes of proteins contain a N-terminal DNA-binding domain (DBD), which also serves as a dimerization interface to other NF-κB transcription factors and, in addition, binds to the inhibitory IκBα protein. The C-terminus of class I proteins contains a number of ankyrin repeats and has transrepression activity. In contrast, the C-terminus of class II proteins has a transactivation function.[1][2][3]NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) is a protein complex that controls the transcription of DNA. NF-κB is found in almost all animal cell types and is involved in cellular responses to stimuli such as stress, cytokines, free radicals, ultraviolet irradiation, oxidized LDL, and bacterial or viral antigens.[1][2][3][4][5] NF-κB plays a key role in regulating the immune response to infection (kappa light chains are critical components of immunoglobulins). Incorrect regulation of NF-κB has been linked to cancer, inflammatory and autoimmune diseases, septic shock, viral infection, and improper immune development. NF-κB has also been implicated in processes of synaptic plasticity and memory.[6][7][8][9][10]
Contents
Discovery
NF-κB was first discovered in the lab of Nobel Prize laureate David Baltimore via its interaction with an 11-base pair sequence in the immunoglobulin light-chain enhancer in B cells.[11]
Structure
All proteins of the NF-κB family share a Rel homology domain in their N-terminus. A subfamily of NF-κB proteins, including RelA, RelB, and c-Rel, have a transactivation domain in their C-termini. In contrast, the NF-κB1 and NF-κB2 proteins are synthesized as large precursors, p105, and p100, which undergo processing to generate the mature NF-κB subunits, p50 and p52, respectively. The processing of p105 and p100 is mediated by the ubiquitin/proteasome pathway and involves selective degradation of their C-terminal region containing ankyrin repeats. Whereas the generation of p52 from p100 is a tightly-regulated process, p50 is produced from constitutive processing of p105.[12][13]
Members
NF-κB family members share structural homology with the retroviral oncoprotein v-Rel, resulting in their classification as NF-κB/Rel proteins.[1]
There are five proteins in the mammalian NF-κB family:[14]
Class Protein Aliases Gene I NF-κB1 p105 → p50 NFKB1 NF-κB2 p100 → p52 NFKB2 II RelA p65 RELA RelB RELB c-Rel REL Below are the five human NF-κB family members:
NFKB1 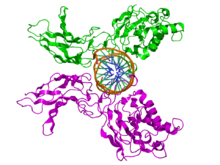
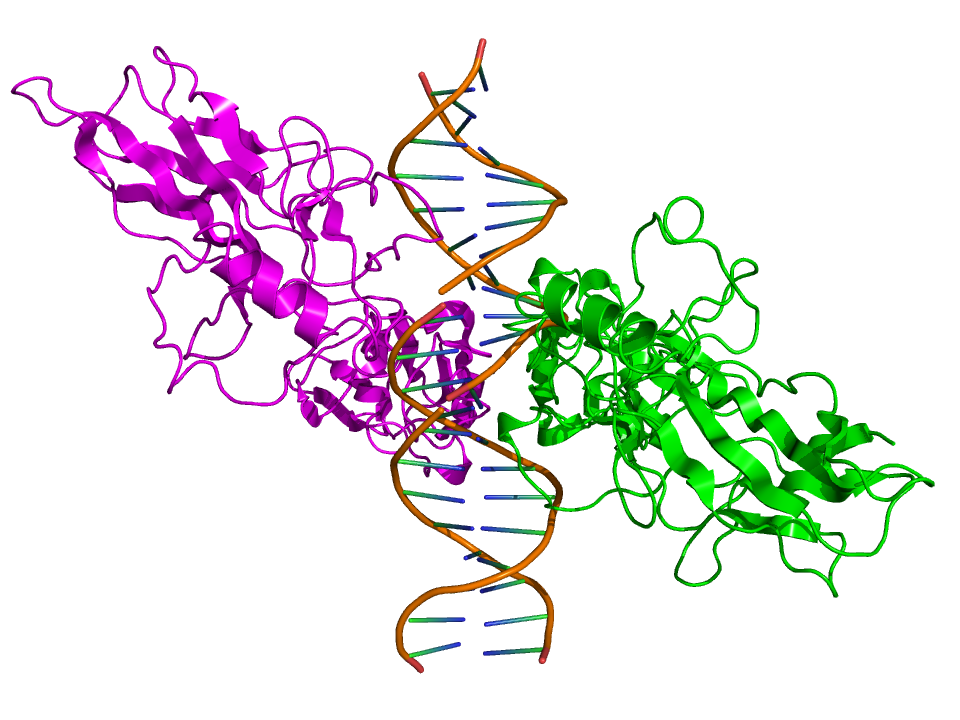
Top view of the crystallographic structure (PDB 1SVC) of a homodimer of the NFKB1 protein (green and magenta) bound to DNA (brown). Identifiers Symbol NFKB1 Entrez 4790 HUGO 7794 OMIM 164011 RefSeq NM_003998 UniProt P19838 Other data Locus Chr. 4 q24 RELA 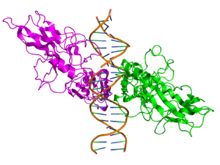
Side view of the crystallographic structure (PDB 2RAM) of a homodimer of the RELA protein (green and magenta) bound to DNA (brown). Identifiers Symbol RELA Entrez 5970 HUGO 9955 OMIM 164014 RefSeq NM_021975 UniProt Q04206 Other data Locus Chr. 11 q13 NFKB2 Identifiers Symbol NFKB2 Entrez 4791 HUGO 7795 OMIM 164012 RefSeq NM_002502 UniProt Q00653 Other data Locus Chr. 10 q24 RELB Identifiers Symbol RELB Entrez 5971 HUGO 9956 OMIM 604758 RefSeq NM_006509 UniProt Q01201 Other data Locus Chr. 19 q13.2-19q13 REL Identifiers Symbol REL Entrez 5966 HUGO 9954 OMIM 164910 RefSeq NM_002908 UniProt Q04864 Other data Locus Chr. 2 p13-p12 Species distribution and evolution
In addition to mammals, NF-κB is found in a number of simple animals as well.[15] These include cnidarians (such as sea anemones, coral and hydra), porifera (sponges), the single-celled eukaryote Capsaspora owczarzaki and insects (such as moths, mosquitoes and fruitflies). The sequencing of the genomes of the mosquitoes A. aegypti and A. gambiae, and the fruitfly D. melanogaster has allowed comparative genetic and evolutionary studies on NF-κB. In those insect species, activation of NF-κB is triggered by the Toll pathway (which evolved independently in insects and mammals) and by the Imd (immune deficiency) pathway.[16]
Signaling
Activation
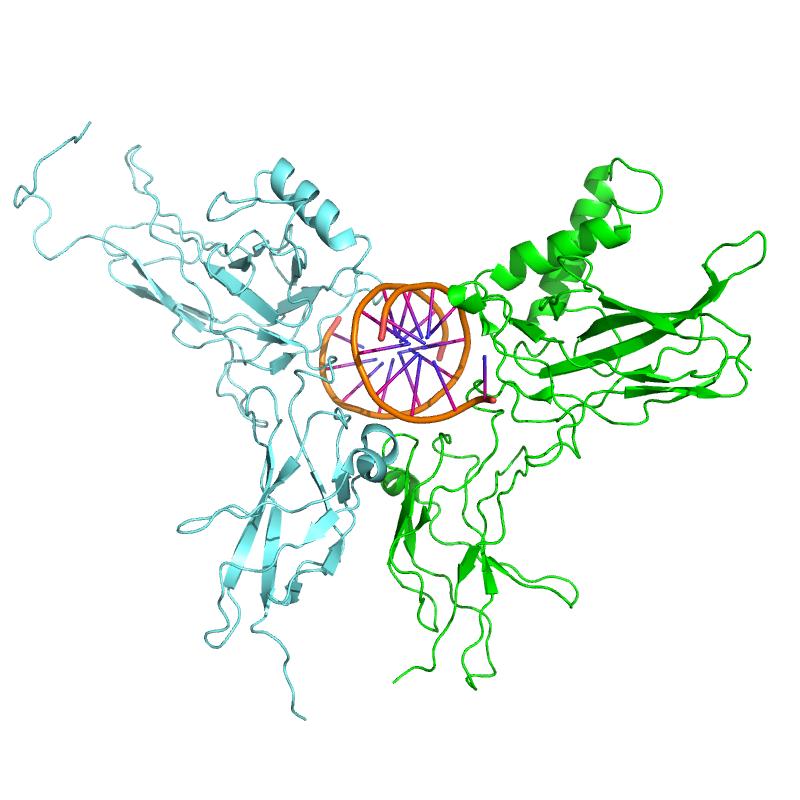
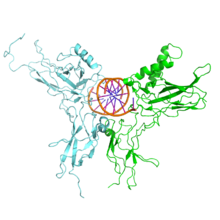 NF-κB (green) heterodimerizes with RelB (cyan) to form a ternary complex with DNA (orange) that promotes gene transcription.[17]
NF-κB (green) heterodimerizes with RelB (cyan) to form a ternary complex with DNA (orange) that promotes gene transcription.[17]NF-κB is important in regulating cellular responses because it belongs to the category of "rapid-acting" primary transcription factors, i.e., transcription factors that are present in cells in an inactive state and do not require new protein synthesis to be activated (other members of this family include transcription factors such as c-Jun, STATs, and nuclear hormone receptors). This allows NF-κB to be a first responder to harmful cellular stimuli. Known inducers of NF-κB activity are highly variable and include reactive oxygen species (ROS), tumor necrosis factor alpha (TNFα), interleukin 1-beta (IL-1β), bacterial lipopolysaccharides (LPS), isoproterenol, cocaine, and ionizing radiation.[18]
Receptor activator of nuclear factor kappa B (RANK), which is a type of TNFR, is a central activator of NF-κB. Osteoprotegerin (OPG), which is a decoy receptor homolog for RANK ligand, inhibits RANK by binding to RANKL, and, thus, osteoprotegerin is tightly involved in regulating NF-κB activation.[19]
Many bacterial products and stimulation of a wide variety of cell-surface receptors lead to NF-κB activation and fairly rapid changes in gene expression.[1] The identification of Toll-like receptors (TLRs) as specific pattern recognition molecules and the finding that stimulation of TLRs leads to activation of NF-κB improved our understanding of how different pathogens activate NF-κB. For example, studies have identified TLR4 as the receptor for the LPS component of Gram-Negative bacteria.[20] TLRs are key regulators of both innate and adaptive immune responses.[21]
Unlike RelA, RelB, and c-Rel, the p50 and p52 NF-κB subunits do not contain transactivation domains in their C terminal halves. Nevertheless, the p50 and p52 NF-κB members play critical roles in modulating the specificity of NF-κB function. Although homodimers of p50 and p52 are, in general, repressors of κB site transcription, both p50 and p52 participate in target gene transactivation by forming heterodimers with RelA, RelB, or c-Rel.[22] In addition, p50 and p52 homodimers also bind to the nuclear protein Bcl-3, and such complexes can function as transcriptional activators.[23][24][25]
Inhibition
In unstimulated cells, the NF-κB dimers are sequestered in the cytoplasm by a family of inhibitors, called IκBs (Inhibitor of κB), which are proteins that contain multiple copies of a sequence called ankyrin repeats. By virtue of their ankyrin repeat domains, the IκB proteins mask the nuclear localization signals (NLS) of NF-κB proteins and keep them sequestered in an inactive state in the cytoplasm.[26]
IκBs are a family of related proteins that have an N-terminal regulatory domain, followed by six or more ankyrin repeats and a PEST domain near their C terminus. Although the IκB family consists of IκBα, IκBβ, IκBε, and Bcl-3, the best-studied and major IκB protein is IκBα. Due to the presence of ankyrin repeats in their C-terminal halves, p105 and p100 also function as IκB proteins. The c-terminal half of p100, that is often referred to as IκBδ, also functions as an inhibitor.[27][28] IκBδ degradation in response to developmental stimuli, such as those transduced through LTβR, potentiate NF-κB dimer activation in a NIK dependent non-canonical pathway.[27][29]
Activation of the NF-κB is initiated by the signal-induced degradation of IκB proteins. This occurs primarily via activation of a kinase called the IκB kinase (IKK). IKK is composed of a heterodimer of the catalytic IKK alpha and IKK beta subunits and a "master" regulatory protein termed NEMO (NF-κB essential modulator) or IKK gamma. When activated by signals, usually coming from the outside of the cell, the IκB kinase phosphorylates two serine residues located in an IκB regulatory domain. When phosphorylated on these serines (e.g., serines 32 and 36 in human IκBα), the IκB inhibitor molecules are modified by a process called ubiquitination, which then leads them to be degraded by a cell structure called the proteasome.
With the degradation of IκB, the NF-κB complex is then freed to enter the nucleus where it can 'turn on' the expression of specific genes that have DNA-binding sites for NF-κB nearby. The activation of these genes by NF-κB then leads to the given physiological response, for example, an inflammatory or immune response, a cell survival response, or cellular proliferation. NF-κB turns on expression of its own repressor, IκBα. The newly synthesized IκBα then re-inhibits NF-κB and, thus, forms an auto feedback loop, which results in oscillating levels of NF-κB activity.[30] In addition, several viruses, including the AIDS virus HIV, have binding sites for NF-κB that controls the expression of viral genes, which in turn contribute to viral replication or viral pathogenicity. In the case of HIV-1, activation of NF-κB may, at least in part, be involved in activation of the virus from a latent, inactive state.[31] YopP is a factor secreted by Yersinia pestis, the causative agent of plague, that prevents the ubiquitination of IκB. This causes this pathogen to effectively inhibit the NF-κB pathway and thus block the immune response of a human infected with Yersinia.[32]
Inhibitors of NF-kB activity
Concerning known protein inhibitors of NF-kB activity, one of them is IFRD1, which represses the activity of NF-kB p65 by enhancing the HDAC-mediated deacetylation of the p65 subunit at lysine 310, by favoring the recruitment of HDAC3 to p65. In fact IFRD1 forms trimolecular complexes with p65 and HDAC3.[33][34]
Non-canonical
A select set of cell-differentiating or developmental stimuli, such as lymphotoxin-α, BAFF or RANKL, activate the non-canonical NF-κB pathway to induce NF-κB/RelB:p52 dimer in the nucleus. In this pathway, activation of the NF-κB inducing kinase (NIK) upon receptor ligation led to the phosphorylation and subsequent proteasomal processing of the NF-κB2 precursor protein p100 into mature p52 subunit in a IKK1/IKKa dependent manner. Then p52 dimerizes with RelB to appear as a nuclear RelB:p52 DNA binding activity and regulate a distinct class of genes.[35] In contrast to the canonical signaling that relies upon NEMO-IKK2 mediated degradation of IκBα, -β, -ε, the non-canonical signaling critically depends on NIK mediated processing of p100 into p52. Given their distinct regulations, these two pathways were thought to be independent of each other. However, recent analyses revealed that synthesis of the constituents of the non-canonical pathway, viz RelB and p52, is controlled by the canonical IKK2-IκB-RelA:p50 signaling.[36] Moreover, generation of the canonical and non-canonical dimers, viz RelA:p50 and RelB:p52, within the cellular milieu are also mechanistically interlinked.[36] These analyses suggest that an integrated NF-κB system network underlies activation of both RelA and RelB containing dimer and that a malfunctioning canonical pathway will lead to an aberrant cellular response also through the non-canonical pathway.
In immunity
NF-κB is a major transcription factor that regulates genes responsible for both the innate and adaptive immune response. Upon activation of either the T- or B-cell receptor, NF-κB becomes activated through distinct signaling components. Upon ligation of the T-cell receptor, protein kinase Lck is recruited and phosphorylates the ITAMs of the CD3 cytoplasmic tail. ZAP70 is then recruited to the phosphorylated ITAMs and helps recruit LAT and PLC-γ, which causes activation of PKC. Through a cascade of phosphorylation events, the kinase complex is activated and NF-κB is able to enter the nucleus to upregulate genes involved in T-cell development, maturation, and proliferation.[37]
In neurons
In addition to roles in mediating cell survival, NF-κB has been demonstrated to have diverse functions in the nervous system including roles in plasticity, learning, and memory. In addition to stimuli that activate NF-κB in other tissues, NF-κB in the nervous system can be activated by Growth Factors (BDNF, NGF) and synaptic transmission such as glutamate.[7] These activators of NF-κB in the nervous system all converge upon the IKK complex and the canonical pathway.
Recently there has been a great deal of interest in the role of NF-κB in the nervous system. Current studies suggest that NF-κB is important for learning and memory in multiple organisms including crabs,[9][10] fruit flies,[38] and mice.[7][8] NF-κB may regulate learning and memory in part by modulating synaptic plasticity,[6][39] synapse function,[38][40][41] as well as by regulating the growth of dendrites[42] and dendritic spines.[41]
Genes that have NF-κB binding sites are shown to have increased expression following learning,[8] suggesting that the transcriptional targets of NF-κB in the nervous system are important for plasticity. Many NF-κB target genes that may be important for plasticity and learning include, glutamate receptors (AMPA-R and NMDA-R),[43][44][45][46] growth factors (BDNF, NGF)[47] cytokines (TNF-alpha, TNFR)[48] kinases (PKAc),[39] and synaptic scaffolding proteins (PSD-95).[41]
Clinical significance
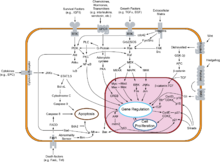 Overview of signal transduction pathways involved in apoptosis.
Overview of signal transduction pathways involved in apoptosis.NF-κB is widely used by eukaryotic cells as a regulator of genes that control cell proliferation and cell survival. As such, many different types of human tumors have misregulated NF-κB: that is, NF-κB is constitutively active. Active NF-κB turns on the expression of genes that keep the cell proliferating and protect the cell from conditions that would otherwise cause it to die via apoptosis.
Defects in NF-κB results in increased susceptibility to apoptosis leading to increased cell death. This is because NF-κB regulates anti-apoptotic genes especially the TRAF1 and TRAF2 and, therefore, checks the activities of the caspase family of enzymes, which are central to most apoptotic processes.[49]
In tumor cells, NF-κB is active either due to mutations in genes encoding the NF-κB transcription factors themselves or in genes that control NF-κB activity (such as IκB genes); in addition, some tumor cells secrete factors that cause NF-κB to become active. Blocking NF-κB can cause tumor cells to stop proliferating, to die, or to become more sensitive to the action of anti-tumor agents. Thus, NF-κB is the subject of much active research among pharmaceutical companies as a target for anti-cancer therapy.[50]
Because NF-κB controls many genes involved in inflammation, it is not surprising that NF-κB is found to be chronically active in many inflammatory diseases, such as inflammatory bowel disease, arthritis, sepsis, gastritis, asthma, atherosclerosis[51] and others. It is important to note that the key regulators of NF-κB are associated with elevated mortality, especially from cardiovascular diseases.[52][53] Elevated NF-κB has also been associated with schizophrenia.[54]
Many natural products (including anti-oxidants) that have been promoted to have anti-cancer and anti-inflammatory activity have also been shown to inhibit NF-κB. There is a controversial US patent (US patent 6,410,516)[55] that applies to the discovery and use of agents that can block NF-κB for therapeutic purposes. This patent is involved in several lawsuits, including Ariad v. Lilly. Recent work by Karin,[56] Ben-Neriah[57] and others has highlighted the importance of the connection between NF-κB, inflammation, and cancer, and underscored the value of therapies that regulate the activity of NF-κB.[58]
Extracts from a number of herbs and dietary plants are efficient inhibitors of NF-kappaB activation in vitro.[59]
As a drug target
Aberrant activation of NF-κB is frequently observed in many cancers. Moreover, suppression of NF-κB limits the proliferation of cancer cells. In addition, NF-κB is a key player in the inflammatory response. Hence methods of inhibiting NF-κB signaling has potential therapeutic application in cancer and inflammatory diseases.[60][61]
The discovery that activation of NF-κB nuclear translocation can be separated from the elevation of oxidant stress[62] gives an important hint to the development of strategies for NF-κB inhibition.
A new drug called denosumab acts to raise bone mineral density and reduce fracture rates in many patient sub-groups by inhibiting RANKL. RANKL acts through its receptor RANK, which in turn promotes NF-κB,[63] RANKL normally works by enabling the differentiation of osteoclasts from monocytes.
Disulfiram, olmesartan and dithiocarbamates can inhibit the nuclear factor-κB (NF-κB) signaling cascade.[64]
Anatabine alleged antiinflammatory effects is claimed to result from modulation of NF-κB activity.[65]
See also
References
- ^ a b c d e Gilmore TD (2006). "Introduction to NF-κB: players, pathways, perspectives". Oncogene 25 (51): 6680–4. doi:10.1038/sj.onc.1209954. PMID 17072321.
- ^ a b c Brasier AR (2006). "The NF-κB regulatory network". Cardiovasc. Toxicol. 6 (2): 111–30. doi:10.1385/CT:6:2:111. PMID 17303919.
- ^ a b c Perkins ND (January 2007). "Integrating cell-signalling pathways with NF-κB and IKK function". Nat. Rev. Mol. Cell Biol. 8 (1): 49–62. doi:10.1038/nrm2083. PMID 17183360.
- ^ Gilmore TD (1999). "The Rel/NF-κB signal transduction pathway: introduction". Oncogene 18 (49): 6842–4. doi:10.1038/sj.onc.1203237. PMID 10602459.
- ^ Tian B, Brasier AR (2003). "Identification of a nuclear factor κ B-dependent gene network". Recent Prog. Horm. Res. 58: 95–130. doi:10.1210/rp.58.1.95. PMID 12795416.
- ^ a b Albensi BC, Mattson MP (2000). "Evidence for the involvement of TNF and NF-κB in hippocampal synaptic plasticity". Synapse 35 (2): 151–9. doi:10.1002/(SICI)1098-2396(200002)35:2<151::AID-SYN8>3.0.CO;2-P. PMID 10611641.
- ^ a b c Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D (October 2003). "NF-kappa B functions in synaptic signaling and behavior". Nat. Neurosci. 6 (10): 1072–8. doi:10.1038/nn1110. PMID 12947408.
- ^ a b c Levenson JM, Choi S, Lee SY, Cao YA, Ahn HJ, Worley KC, Pizzi M, Liou HC, Sweatt JD (April 2004). "A bioinformatics analysis of memory consolidation reveals involvement of the transcription factor c-rel". J. Neurosci. 24 (16): 3933–43. doi:10.1523/JNEUROSCI.5646-03.2004. PMID 15102909.
- ^ a b Freudenthal R, Locatelli F, Hermitte G, Maldonado H, Lafourcade C, Delorenzi A, Romano A (February 1998). "Kappa-B like DNA-binding activity is enhanced after spaced training that induces long-term memory in the crab Chasmagnathus". Neurosci. Lett. 242 (3): 143–6. doi:10.1016/S0304-3940(98)00059-7. PMID 9530926.
- ^ a b Merlo E, Freudenthal R, Romano A (2002). "The IkappaB kinase inhibitor sulfasalazine impairs long-term memory in the crab Chasmagnathus". Neuroscience 112 (1): 161–72. doi:10.1016/S0306-4522(02)00049-0. PMID 12044481.
- ^ Sen R, Baltimore D (1986). "Multiple nuclear factors interact with the immunoglobulin enhancer sequences". Cell 46 (5): 705–16. doi:10.1016/0092-8674(86)90346-6. PMID 3091258.
- ^ Karin M, Ben-Neriah Y (2000). "Phosphorylation meets ubiquitination: the control of NF-κB activity". Annu. Rev. Immunol. 18: 621–63. doi:10.1146/annurev.immunol.18.1.621. PMID 10837071.
- ^ Senftleben U, Cao Y, Xiao G, Greten FR, Krähn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun SC, Karin M (2001). "Activation by IKKalpha of a second, evolutionary conserved, NF-κB signaling pathway". Science 293 (5534): 1495–9. doi:10.1126/science.1062677. PMID 11520989.
- ^ Nabel GJ, Verma IM (November 1993). "Proposed NF-κB/IκB family nomenclature". Genes Dev. 7 (11): 2063. doi:10.1101/gad.7.11.2063. PMID 8224837.
- ^ Ghosh S, May MJ, Kopp EB (1998). "NF-κB and Rel proteins: evolutionarily conserved mediators of immune responses". Annu. Rev. Immunol. 16: 225–60. doi:10.1146/annurev.immunol.16.1.225. PMID 9597130.
- ^ Waterhouse RM, Kriventseva EV, Meister S, Xi Z, Alvarez KS, Bartholomay LC, Barillas-Mury C, Bian G, Blandin S, Christensen BM, Dong Y, Jiang H, Kanost MR, Koutsos AC, Levashina EA, Li J, Ligoxygakis P, Maccallum RM, Mayhew GF, Mendes A, Michel K, Osta MA, Paskewitz S, Shin SW, Vlachou D, Wang L, Wei W, Zheng L, Zou Z, Severson DW, Raikhel AS, Kafatos FC, Dimopoulos G, Zdobnov EM, Christophides GK (2007). "Evolutionary dynamics of immune-related genes and pathways in disease-vector mosquitoes". Science 316 (5832): 1738–43. doi:10.1126/science.1139862. PMC 2042107. PMID 17588928. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2042107.
- ^ PDB 3do7; Fusco AJ, Huang DB, Miller D, Wang VY, Vu D, Ghosh G (February 2009). "NF-kappaB p52:RelB heterodimer recognizes two classes of kappaB sites with two distinct modes". EMBO Rep. 10 (2): 152–9. doi:10.1038/embor.2008.227. PMC 2637311. PMID 19098713. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2637311.
- ^ (a) Chandel NS, Trzyna WC, McClintock DS, Schumacker PT (July 2000). "Role of oxidants in NF-kappa B activation and TNF-alpha gene transcription induced by hypoxia and endotoxin". J Immunol 165 (2): 1013–1021. PMID 10878378.; (b) Fitzgerald DC, Meade KG, McEvoy AN, Lillis L, Murphy EP, MacHugh DE, Baird AW (March 2007). "Tumour necrosis factor-alpha (TNF-alpha) increases nuclear factor kappaB (NFkappaB) activity in and interleukin-8 (IL-8) release from bovine mammary epithelial cells". Vet Immunol Immunopathol 116 (1-2): 59–68. doi:10.1016/j.vetimm.2006.12.008. PMID 17276517.; (c) Renard P, Zachary MD, Bougelet C, Mirault ME, Haegeman G, Remacle J, Raes M (January 1997). "Effects of antioxidant enzyme modulations on interleukin-1-induced nuclear factor kappa B activation". Biochem Pharmacol 53 (2): 149–160. PMID 9037247.; (d) Qin H, Wilson CA, Lee SJ, Zhao X, Benveniste EN (November 2005). "LPS induces CD40 gene expression through the activation of NF-kappaB and STAT-1alpha in macrophages and microglia". Blood 106 (9): 3114–3122. doi:10.1182/blood-2005-02-0759. PMC 1895321. PMID 16020513. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1895321.; (e) Takemoto Y, Yoshiyama M, Takeuchi K, Omura T, Komatsu R, Izumi Y, Kim S, Yoshikawa J (November 1999). "Increased JNK, AP-1 and NF-kappa B DNA binding activities in isoproterenol-induced cardiac remodeling". J Mol Cell Cardiol 31 (11): 2017–2030. doi:10.1006/jmcc.1999.1033. PMID 10591028.; (f) Hargrave BY, Tiangco DA, Lattanzio FA, Beebe SJ (2003). "Cocaine, not morphine, causes the generation of reactive oxygen species and activation of NF-kappaB in transiently cotransfected heart cells". Cardiovasc Toxicol 3 (2): 141–151. doi:10.1385/CT:3:2:141. PMID 14501032.; (g) Basu S, Rosenzweig KR, Youmell M, Price BD (June 1998). "The DNA-dependent protein kinase participates in the activation of NF kappa B following DNA damage". Biochem Biophys Res Commun 247 (1): 79–83. doi:10.1006/bbrc.1998.8741. PMID 9636658.
- ^ Baud'huin M, Lamoureux F, Duplomb L, Rédini F, Heymann D (September 2007). "RANKL, RANK, osteoprotegerin: key partners of osteoimmunology and vascular diseases". Cell Mol Life Sci 64 (18): 2334–2350. doi:10.1007/s00018-007-7104-0. PMID 17530461.
- ^ Doyle SL, O'Neill LA (October 2006). "Toll-like receptors: from the discovery of NFκB to new insights into transcriptional regulations in innate immunity". Biochem. Pharmacol. 72 (9): 1102–13. doi:10.1016/j.bcp.2006.07.010. PMID 16930560.
- ^ Hayden MS, West AP, Ghosh S (October 2006). "NF-κB and the immune response". Oncogene 25 (51): 6758–80. doi:10.1038/sj.onc.1209943. PMID 17072327.
- ^ Li Q, Verma IM (2002). "NF-κB regulation in the immune system". Nat. Rev. Immunol. 2 (10): 725–34. doi:10.1038/nri910. PMID 12360211.
- ^ Fujita T, Nolan GP, Liou HC, Scott ML, Baltimore D (1993). "The candidate proto-oncogene bcl-3 encodes a transcriptional coactivator that activates through NF-κB p50 homodimers". Genes Dev. 7 (7B): 1354–63. doi:10.1101/gad.7.7b.1354. PMID 8330739.
- ^ Franzoso G, Bours V, Park S, Tomita-Yamaguchi M, Kelly K, Siebenlist U (1992). "The candidate oncoprotein Bcl-3 is an antagonist of p50/NF-κB-mediated inhibition". Nature 359 (6393): 339–42. doi:10.1038/359339a0. PMID 1406939.
- ^ Bours V, Franzoso G, Azarenko V, Park S, Kanno T, Brown K, Siebenlist U (1993). "The oncoprotein Bcl-3 directly transactivates through κ B motifs via association with DNA-binding p50B homodimers". Cell 72 (5): 729–39. doi:10.1016/0092-8674(93)90401-B. PMID 8453667.
- ^ Jacobs MD, Harrison SC (1998). "Structure of an IκBalpha/NF-κB complex". Cell 95 (6): 749–58. doi:10.1016/S0092-8674(00)81698-0. PMID 9865693.
- ^ a b Basak S, Kim H, Kearns JD, Tergaonkar V, O'dea E, Werner SL, Benedict CA, Ware CF, Ghosh G, Verma IM, Hoffmann A (2007). "A fourth IκB protein within the NF-κB signaling module". Cell 128 (2): 369–81. doi:10.1016/j.cell.2006.12.033. PMC 1831796. PMID 17254973. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1831796..
- ^ Dobrzanski P, Ryseck RP, Bravo R (1995). "Specific inhibition of RelB/p52 transcriptional activity by the C-terminal domain of p100". Oncogene 10 (5): 1003–7. PMID 7898917.
- ^ Lo JC, Basak S, James ES, Quiambo RS, Kinsella MC, Alegre ML, Weih F, Franzoso G, Hoffmann A, Fu YX (2006). "Coordination between NF-κB family members p50 and p52 is essential for mediating LTbetaR signals in the development and organization of secondary lymphoid tissues". Blood 107 (3): 1048–55. doi:10.1182/blood-2005-06-2452. PMC 1895903. PMID 16195333. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1895903.
- ^ Nelson DE, Ihekwaba AE, Elliott M, Johnson JR, Gibney CA, Foreman BE, Nelson G, See V, Horton CA, Spiller DG, Edwards SW, McDowell HP, Unitt JF, Sullivan E, Grimley R, Benson N, Broomhead D, Kell DB, White MR (2004). "Oscillations in NF-κB signaling control the dynamics of gene expression". Science 306 (5696): 704–8. doi:10.1126/science.1099962. PMID 15499023.
- ^ Hiscott J, Kwon H, Génin P (January 2001). "Hostile takeovers: viral appropriation of the NF-κB pathway". J. Clin. Invest. 107 (2): 143–51. doi:10.1172/JCI11918. PMC 199181. PMID 11160127. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=199181.
- ^ Adkins I, Schulz S, Borgmann S, Autenrieth IB, Gröbner S (February 2008). "Differential roles of Yersinia outer protein P-mediated inhibition of nuclear factor-κB in the induction of cell death in dendritic cells and macrophages". J. Med. Microbiol. 57 (Pt 2): 139–44. doi:10.1099/jmm.0.47437-0. PMID 18201977.
- ^ Micheli L, Leonardi L, Conti F, Buanne P, Canu N, Caruso M, Tirone F (March 2005). "PC4 coactivates MyoD by relieving the histone deacetylase 4-mediated inhibition of myocyte enhancer factor 2C". Mol. Cell. Biol. 25 (6): 2242–59. doi:10.1128/MCB.25.6.2242-2259.2005. PMC 1061592. PMID 15743821. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1061592.
- ^ Micheli L, Leonardi L, Conti F, Maresca G, Colazingari S, Mattei E, Lira SA, Farioli-Vecchioli S, Caruso M, Tirone F (February 2011). "PC4/Tis7/IFRD1 stimulates skeletal muscle regeneration and is involved in myoblast differentiation as a regulator of MyoD and NF-kappaB". J. Biol. Chem. 286 (7): 5691–707. doi:10.1074/jbc.M110.162842. PMC 3037682. PMID 21127072. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3037682.
- ^ Bonizzi G, Bebien M, Otero DC, Johnson-Vroom KE, Cao Y, Vu D, Jegga AG, Aronow BJ, Ghosh G, Rickert RC, Karin M (2004). "Activation of IKKalpha target genes depends on recognition of specific kappaB binding sites by RelB:p52 dimers". EMBO J 23 (21): 4202–10. doi:10.1038/sj.emboj.7600391. PMID 11547050.
- ^ a b Basak S, Shih VF, Hoffmann A (2008). "Generation and activation of multiple dimeric transcription factors within the NF-kappaB signaling system". Mol Cell Biol 28 (10): 3139–50. doi:10.1128/MCB.01469-07. PMC 2423155. PMID 18299388. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2423155.
- ^ Livolsi A, Busuttil V, Imbert V, Abraham RT, Peyron JF (March 2001). "Tyrosine phosphorylation-dependent activation of NF-κB. Requirement for p56 LCK and ZAP-70 protein tyrosine kinases". Eur. J. Biochem. 268 (5): 1508–15. doi:10.1046/j.1432-1327.2001.02028.x. PMID 11231305.
- ^ a b Heckscher ES, Fetter RD, Marek KW, Albin SD, Davis GW (September 2007). "NF-kappaB, IkappaB, and IRAK control glutamate receptor density at the Drosophila NMJ". Neuron 55 (6): 859–73. doi:10.1016/j.neuron.2007.08.005. PMC 2701504. PMID 17880891. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2701504.
- ^ a b Kaltschmidt B, Ndiaye D, Korte M, Pothion S, Arbibe L, Prüllage M, Pfeiffer J, Lindecke A, Staiger V, Israël A, Kaltschmidt C, Mémet S (April 2006). "NF-kappaB regulates spatial memory formation and synaptic plasticity through protein kinase A/CREB signaling". Mol. Cell. Biol. 26 (8): 2936–46. doi:10.1128/MCB.26.8.2936-2946.2006. PMC 1446931. PMID 16581769. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1446931.
- ^ Wang J, Fu XQ, Lei WL, Wang T, Sheng AL, Luo ZG (August 2010). "Nuclear factor kappaB controls acetylcholine receptor clustering at the neuromuscular junction". J. Neurosci. 30 (33): 11104–13. doi:10.1523/JNEUROSCI.2118-10.2010. PMID 20720118.
- ^ a b c Boersma MC, Dresselhaus EC, De Biase LM, Mihalas AB, Bergles DE, Meffert MK (April 2011). "A Requirement for Nuclear Factor-{kappa}B in Developmental and Plasticity-Associated Synaptogenesis". J. Neurosci. 31 (14): 5414–25. doi:10.1523/JNEUROSCI.2456-10.2011. PMID 21471377.
- ^ Gutierrez H, Hale VA, Dolcet X, Davies A (April 2005). "NF-kappaB signalling regulates the growth of neural processes in the developing PNS and CNS". Development 132 (7): 1713–26. doi:10.1242/dev.01702. PMID 15743881.
- ^ Borges K, Dingledine R (July 2001). "Functional organization of the GluR1 glutamate receptor promoter". J. Biol. Chem. 276 (28): 25929–38. doi:10.1074/jbc.M009105200. PMID 11340067.
- ^ Yu Z, Cheng G, Wen X, Wu GD, Lee WT, Pleasure D (October 2002). "Tumor necrosis factor alpha increases neuronal vulnerability to excitotoxic necrosis by inducing expression of the AMPA-glutamate receptor subunit GluR1 via an acid sphingomyelinase- and NF-kappaB-dependent mechanism". Neurobiol. Dis. 11 (1): 199–213. doi:10.1006/nbdi.2002.0530. PMID 12460558.
- ^ Richter M, Suau P, Ponte I (July 2002). "Sequence and analysis of the 5' flanking and 5' untranslated regions of the rat N-methyl-D-aspartate receptor 2A gene". Gene 295 (1): 135–42. doi:10.1016/S0378-1119(02)00833-8. PMID 12242020.
- ^ Begni S, Moraschi S, Bignotti S, Fumagalli F, Rillosi L, Perez J, Gennarelli M (April 2003). "Association between the G1001C polymorphism in the GRIN1 gene promoter region and schizophrenia". Biol. Psychiatry 53 (7): 617–9. doi:10.1016/S0006-3223(02)01783-3. PMID 12679240.
- ^ Zaheer A, Yorek MA, Lim R (December 2001). "Effects of glia maturation factor overexpression in primary astrocytes on MAP kinase activation, transcription factor activation, and neurotrophin secretion". Neurochem. Res. 26 (12): 1293–9. doi:10.1023/A:1014241300179. PMID 11885780.
- ^ Qiu J, Hu X, Nesic O, Grafe MR, Rassin DK, Wood TG, Perez-Polo JR (July 2004). "Effects of NF-kappaB oligonucleotide "decoys" on gene expression in P7 rat hippocampus after hypoxia/ischemia". J. Neurosci. Res. 77 (1): 108–18. doi:10.1002/jnr.20156. PMID 15197744.
- ^ Sheikh MS, Huang Y (2003). "Death receptor activation complexes: it takes two to activate TNF receptor 1". Cell Cycle 2 (6): 550–2. PMID 14504472. http://www.landesbioscience.com/journals/cc/article/566/.
- ^ Escárcega RO, Fuentes-Alexandro S, García-Carrasco M, Gatica A, Zamora A (2007). "The transcription factor nuclear factor-κB and cancer". Clinical Oncology (Royal College of Radiologists (Great Britain)) 19 (2): 154–61. doi:10.1016/j.clon.2006.11.013. PMID 17355113.
- ^ Monaco C, Andreakos E, Kiriakidis S, Mauri C, Bicknell C, Foxwell B, Cheshire N, Paleolog E, Feldmann M. Canonical pathway of nuclear factor kappa B activation selectively regulates proinflammatory and prothrombotic responses in human atherosclerosis. Proc Natl Acad Sci U S A. 2004 Apr 13;101(15):5634-9. Epub 2004 Apr 2. PubMed PMID: 15064395 Link to paper;; PubMed Central PMCID: PMC397455.
- ^ Venuraju SM, Yerramasu A, Corder R, Lahiri A (May 2010). "Osteoprotegerin as a predictor of coronary artery disease and cardiovascular mortality and morbidity". J. Am. Coll. Cardiol. 55 (19): 2049–61. doi:10.1016/j.jacc.2010.03.013. PMID 20447527.
- ^ Lieb W, Gona P, Larson MG, Massaro JM, Lipinska I, Keaney JF, Rong J, Corey D, Hoffmann U, Fox CS, Vasan RS, Benjamin EJ, O'Donnell CJ, Kathiresan S (September 2010). "Biomarkers of the osteoprotegerin pathway: clinical correlates, subclinical disease, incident cardiovascular disease, and mortality". Arterioscler. Thromb. Vasc. Biol. 30 (9): 1849–54. doi:10.1161/ATVBAHA.109.199661. PMC 3039214. PMID 20448212. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3039214.
- ^ Song XQ, Lv LX, Li WQ, Hao YH, Zhao JP (March 2009). "The interaction of nuclear factor-kappa B and cytokines is associated with schizophrenia". Biol. Psychiatry 65 (6): 481–8. doi:10.1016/j.biopsych.2008.10.018. PMID 19058794.
- ^ US patent 6410516, Baltimore D; Sen R; Sharp PA; Singh H; Staudt L; Lebowitz JH; Baldwin Jr AS; Clerc RG; Corcoran LM; Baeuerle PA; Lenardo MJ; Fan C-M; Maniatis TPD, "Nuclear factors associated with transcriptional regulation", issued 2002-06-25
- ^ Karin M (March 2008). "The IκB kinase - a bridge between inflammation and cancer". Cell Res. 18 (3): 334–42. doi:10.1038/cr.2008.30. PMID 18301380.
- ^ Pikarsky E, Ben-Neriah Y (April 2006). "NF-κB inhibition: a double-edged sword in cancer?". Eur. J. Cancer 42 (6): 779–84. doi:10.1016/j.ejca.2006.01.011. PMID 16530406.
- ^ Mantovani A, Marchesi F, Portal C, Allavena P, Sica A (2008). "Linking inflammation reactions to cancer: novel targets for therapeutic strategies". Adv. Exp. Med. Biol. 610: 112–27. doi:10.1007/978-0-387-73898-7_9. PMID 18593019.
- ^ Paur I, Balstad TR, Kolberg M, Pedersen MK, Austenaa LM, Jacobs DR, Blomhoff R (May 2010). "Extract of oregano, coffee, thyme, clove, and walnuts inhibits NF-kappaB in monocytes and in transgenic reporter mice". Cancer Prev Res (Phila) 3 (5): 653–63. doi:10.1158/1940-6207.CAPR-09-0089. PMID 20424131.
- ^ Garg A, Aggarwal BB (June 2002). "Nuclear transcription factor-kappaB as a target for cancer drug development". Leukemia 16 (6): 1053–68. doi:10.1038/sj.leu.2402482. PMID 12040437.
- ^ Sethi G, Sung B, Aggarwal BB (January 2008). "Nuclear factor-kappaB activation: from bench to bedside". Exp. Biol. Med. (Maywood) 233 (1): 21–31. doi:10.3181/0707-MR-196. PMID 18156302.
- ^ Vlahopoulos S, Boldogh I, Casola A, Brasier AR (September 1999). "Nuclear factor-κB-dependent induction of interleukin-8 gene expression by tumor necrosis factor alpha: evidence for an antioxidant sensitive activating pathway distinct from nuclear translocation". Blood 94 (6): 1878–89. PMID 10477716. http://bloodjournal.hematologylibrary.org/cgi/content/abstract/bloodjournal;94/6/1878.
- ^ Hamdy NA (2008). "Denosumab: RANKL inhibition in the management of bone loss". Drugs Today (Barc). 44 (1): 7–21. doi:10.1358/dot.2008.44.1.1178467. PMID 18301800.
- ^ Cvek B, Dvorak Z (2007). "Targeting of nuclear factor-κB and proteasome by dithiocarbamate complexes with metals". Curr. Pharm. Des. 13 (30): 3155–67. doi:10.2174/138161207782110390. PMID 17979756.
- ^ "Role of RCP006 as an anti-inflammatory agent". Roskamp Institute. http://www.rfdn.org/inflammaging3.html. Retrieved 2011-09-06.
External links
- MeSH NF-kappa+B
- Sankar Ghosh (2006). Handbook of Transcription Factor NF-κB. Boca Raton: CRC. ISBN 0-8493-2794-6.
- Thomas D Gilmore. "The Rel/NF-κB Signal Transduction Pathway". Boston University. http://people.bu.edu/gilmore/nf-kb/. Retrieved 2007-12-02.
Transcription factors and intracellular receptors (1) Basic domains (1.1) Basic leucine zipper (bZIP)Activating transcription factor (AATF, 1, 2, 3, 4, 5, 6, 7) · AP-1 (c-Fos, FOSB, FOSL1, FOSL2, JDP2, c-Jun, JUNB, JUND) · BACH (1, 2) · BATF · BLZF1 · C/EBP (α, β, γ, δ, ε, ζ) · CREB (1, 3, L1) · CREM · DBP · DDIT3 · GABPA · HLF · MAF (B, F, G, K) · NFE (2, L1, L2, L3) · NFIL3 · NRL · NRF (1, 2, 3) · XBP1(1.2) Basic helix-loop-helix (bHLH)ATOH1 · AhR · AHRR · ARNT · ASCL1 · BHLHB2 · BMAL (ARNTL, ARNTL2) · CLOCK · EPAS1 · FIGLA · HAND (1, 2) · HES (5, 6) · HEY (1, 2, L) · HES1 · HIF (1A, 3A) · ID (1, 2, 3, 4) · LYL1 · MESP2 · MXD4 · MYCL1 · MYCN · Myogenic regulatory factors (MyoD, Myogenin, MYF5, MYF6) · Neurogenins (1, 2, 3) · NeuroD (1, 2) · NPAS (1, 2, 3) · OLIG (1, 2) · Pho4 · Scleraxis · SIM (1, 2) · TAL (1, 2) · Twist · USF1(1.3) bHLH-ZIP(1.4) NF-1(1.5) RF-X(1.6) Basic helix-span-helix (bHSH)(2) Zinc finger DNA-binding domains (2.1) Nuclear receptor (Cys4)subfamily 1 (Thyroid hormone (α, β), CAR, FXR, LXR (α, β), PPAR (α, β/δ, γ), PXR, RAR (α, β, γ), ROR (α, β, γ), Rev-ErbA (α, β), VDR)
subfamily 2 (COUP-TF (I, II), Ear-2, HNF4 (α, γ), PNR, RXR (α, β, γ), Testicular receptor (2, 4), TLX)
subfamily 3 (Steroid hormone (Androgen, Estrogen (α, β), Glucocorticoid, Mineralocorticoid, Progesterone), Estrogen related (α, β, γ))
subfamily 4 NUR (NGFIB, NOR1, NURR1) · subfamily 5 (LRH-1, SF1) · subfamily 6 (GCNF) · subfamily 0 (DAX1, SHP)(2.2) Other Cys4(2.3) Cys2His2General transcription factors (TFIIA, TFIIB, TFIID, TFIIE (1, 2), TFIIF (1, 2), TFIIH (1, 2, 4, 2I, 3A, 3C1, 3C2))
ATBF1 · BCL (6, 11A, 11B) · CTCF · E4F1 · EGR (1, 2, 3, 4) · ERV3 · GFI1 · GLI-Krüppel family (1, 2, 3, REST, S2, YY1) · HIC (1, 2) · HIVEP (1, 2, 3) · IKZF (1, 2, 3) · ILF (2, 3) · KLF (2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 17) · MTF1 · MYT1 · OSR1 · PRDM9 · SALL (1, 2, 3, 4) · SP (1, 2, 4, 7, 8) · TSHZ3 · WT1 · Zbtb7 (7A, 7B) · ZBTB (16, 17, 20, 32, 33, 40) · zinc finger (3, 7, 9, 10, 19, 22, 24, 33B, 34, 35, 41, 43, 44, 51, 74, 143, 146, 148, 165, 202, 217, 219, 238, 239, 259, 267, 268, 281, 295, 300, 318, 330, 346, 350, 365, 366, 384, 423, 451, 452, 471, 593, 638, 644, 649, 655)(2.4) Cys6(2.5) Alternating composition(3) Helix-turn-helix domains (3.1) HomeodomainARX · CDX (1, 2) · CRX · CUTL1 · DBX (1, 2) · DLX (3, 4, 5) · EMX2 · EN (1, 2) · FHL (1, 2, 3) · HESX1 · HHEX · HLX · Homeobox (A1, A2, A3, A4, A5, A7, A9, A10, A11, A13, B1, B2, B3, B4, B5, B6, B7, B8, B9, B13, C4, C5, C6, C8, C9, C10, C11, C12, C13, D1, D3, D4, D8, D9, D10, D11, D12, D13) · HOPX · IRX (1, 2, 3, 4, 5, 6, MKX) · LMX (1A, 1B) · MEIS (1, 2) · MEOX2 · MNX1 · MSX (1, 2) · NANOG · NKX (2-1, 2-2, 2-3, 2-5, 3-1, 3-2, 6-1, 6-2) · NOBOX · PBX (1, 2, 3) · PHF (1, 3, 6, 8, 10, 16, 17, 20, 21A) · PHOX (2A, 2B) · PITX (1, 2, 3) · POU domain (PIT-1, BRN-3: A, B, C, Octamer transcription factor: 1, 2, 3/4, 6, 7, 11) · OTX (1, 2) · PDX1 · SATB2 · SHOX2 · VAX1 · ZEB (1, 2)(3.2) Paired box(3.3) Fork head / winged helix(3.4) Heat Shock Factors(3.5) Tryptophan clusters(3.6) TEA domain(4) β-Scaffold factors with minor groove contacts (4.1) Rel homology region(4.2) STAT(4.3) p53(4.4) MADS box(4.6) TATA binding proteins(4.7) High-mobility group(4.10) Cold-shock domainCSDA, YBX1(4.11) Runt(0) Other transcription factors (0.2) HMGI(Y)(0.3) Pocket domain(0.6) MiscellaneousCategories:- Genes on chromosome 4
- Genes on chromosome 11
- Genes on chromosome 10
- Genes on chromosome 19
- Genes on chromosome 2
- Protein complexes
- Programmed cell death
- Transcription factors


Wikimedia Foundation. 2010.