- Cyclin-dependent kinase
-
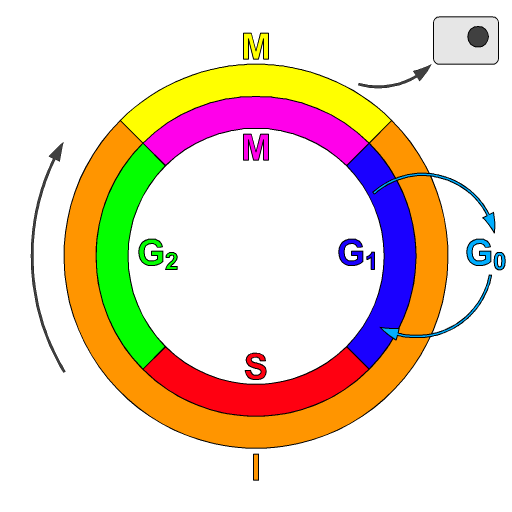
 Schematic of the cell cycle. outer ring: I=Interphase, M=Mitosis; inner ring: M=Mitosis; G1=Gap phase 1; S=Synthesis; G2=Gap phase 2. The duration of mitosis in relation to the other phases has been exaggerated in this diagram
Schematic of the cell cycle. outer ring: I=Interphase, M=Mitosis; inner ring: M=Mitosis; G1=Gap phase 1; S=Synthesis; G2=Gap phase 2. The duration of mitosis in relation to the other phases has been exaggerated in this diagram
Cyclin-dependent kinases (CDKs) are a family of protein kinases first discovered for their role in regulating the cell cycle. They are also involved in regulating transcription, mRNA processing, and the differentiation of nerve cells.[1] They are present in all known eukaryotes, and their regulatory function in the cell cycle has been evolutionarily conserved. In fact, yeast cells can proliferate normally when their CDK gene has been replaced with the homologous human gene.[1][2] CDKs are relatively small proteins, with molecular weights ranging from 34 to 40 kDa, and contain little more than the kinase domain.[1] By definition, a CDK binds a regulatory protein called a cyclin. Without cyclin, CDK has little kinase activity; only the cyclin-CDK complex is an active kinase. CDKs phosphorylate their substrates on serines and threonines, so they are serine-threonine kinases.[1] The consensus sequence for the phosphorylation site in the amino acid sequence of a CDK substrate is [S/T*]PX[K/R], where S/T* is the phosphorylated serine or threonine, P is proline, X is any amino acid, K is lysine, and R is arginine [1]
Contents
Types
Table 1: Known CDKs, their cyclin partners, and their functions in the human [3] and consequences of deletion in mice.[4]
CDK Cyclin partner Function Deletion Phenotype in Mice Cdk1 Cyclin B M phase None. ~E2.5. Cdk2 Cyclin E G1/S transition Reduced size, imparted neural progenitor cell proliferation. Viable, but both males & females sterile. Cdk2 Cyclin A S phase, G2 phase Cdk3 Cyclin C G1 phase ? No defects. Viable, fertile. Cdk4 Cyclin D G1 phase Reduced size, insulin deficient diabetes. Viable, but both male & female infertile. Cdk5 p35 Transcription Severe neurological defects. Died immediately after birth. Cdk6 Cyclin D G1 phase Cdk7 Cyclin H CDK-activating kinase, transcription Cdk8 Cyclin C Transcription Cdk11 Cyclin L ? Mitotic defects. E3.5. ? Cyclin F ? Defects in extraembryonic tissues. E10.5. ? Cyclin G ? CDKs and Cyclins in the Cell Cycle
Most of the known cyclin-CDK complexes regulate the progression through the cell cycle. Animal cells contain at least nine CDKs, four of which, Cdk1, 2, 3, and 4, are directly involved in cell cycle regulation.[1] In mammalian cells, CDK1, with its partners cyclin A2 and B1, alone can drive the cell cycle.[4] Another one, Cdk5, is involved indirectly as the CDK-activating kinase.[1] Cyclin-CDK complexes phosphorylate substrates appropriate for the particular cell cycle phase.[3] Cyclin-CDK complexes in earlier cell-cycle phase help activate cyclin-CDK complexes in later phases.[1]
Table 2: Cyclins and CDKs by Cell-Cycle Phase
Phase Cyclin CDK G0 C Cdk3 G1 D, E Cdk4, Cdk2, Cdk6 S A, E Cdk2 G2 A Cdk2, Cdk1 M B Cdk1 Table 3: Cyclin-dependent kinases that control the cell cycle in model organisms.[1]
Species Name Original name Size (amino acids) Function Saccharomyces cerevisiae Cdk1 Cdc28 298 All cell-cycle stages Schizosaccharomyces pombe Cdk1 Cdc2 297 All cell-cycle stages Drosophila melanogaster Cdk1 Cdc2 297 M Cdk2 Cdc2c 314 G1/S, S, possibly M Cdk4 Cdk4/6 317 G1, promotes growth Xenopus laevis Cdk1 Cdc2 301 M Cdk2 297 S, possibly M Homo sapiens Cdk1 Cdc2 297 M Cdk2 298 G1, S, possibly M Cdk4 301 G1 Cdk6 326 G1 A list of CDKs with their regulator protein, cyclin or other.
- CDK1; cyclin A, cyclin B
- CDK2; cyclin A, cyclin E
- CDK3; cyclin C
- CDK4; cyclin D1, cyclin D2, cyclin D3
- CDK5; CDK5R1, CDK5R2. See also CDKL5.
- CDK6; cyclin D1, cyclin D2, cyclin D3
- CDK7; cyclin H
- CDK8; cyclin C
- CDK9; cyclin T1, cyclin T2a, cyclin T2b, cyclin K
- CDK10
- CDK11 (CDC2L2) ; cyclin L
- CDK12 (CRKRS) ; cyclin L
- CDK13 (CDC2L5) ; cyclin L
Regulation of CDK activity
CDK levels remains relatively constant throughout the cell cycle and most regulation is post-translational. Most knowledge of CDK structure and function is based on CDKs of S. pombe (Cdc2), S. cerevisiae (CDC28), and vertebrates (CDC2 and CDK2). The four major mechanisms of CDK regulation are cyclin binding, CAK phosphorylation, regulatory inhibitory phosphorylation, and binding of CDK inhibitory subunits (CKIs).[5]
Cyclin binding
The active site, or ATP-binding site, of all kinases is a cleft between a small amino-terminal lobe and a larger carboxy-terminal lobe.[1] The structure of human Cdk2 revealed that CDKs have a modified ATP-binding site that can be regulated by cyclin binding.[1] Phosphorylation by CDK-activating kinase (CAK) at Thr 161 on the T-loop increases the complex activity. Without cyclin, a flexible loop called the activation loop or T-loop blocks the cleft, and the position of several key amino acid residues is not optimal for ATP-binding.[1] With cyclin, two alpha helices change position to permit ATP binding. One of them, the L12 helix that comes just before the T-loop in the primary sequence, becomes a beta strand and helps rearrange the T-loop, so it no longer blocks the active site.[1] The other alpha helix called the PSTAIRE helix rearranges and helps changes the position of the key amino acid residues in the active site.[1]
There is considerable specificity in which cyclin binds with CDK.[3] Furthermore, cyclin binding determines the specificity of the cyclin-CDK complex for particular substrates.[3] Cyclins can directly bind the substrate or localize the Cdk to a subcellular area where the substrate is found. Substrate specificity of S cyclins is imparted by the hydrophobic batch (centered on the MRAIL sequence), which has affinity for substrate proteins that contain a hydrophobic RXL (or Cy) motif. Cyclin B1 and B2 can localize Cdk1 to the nucleus and the Golgi, respectively, through a localization sequence outside the Cdk-binding region.[1]
Phosphorylation
Full kinase activity requires an activating phosphorylation on a threonine adjacent to the active site.[1] The identity of the CDK-activating kinase (CAK) that performs this phosphorylation varies across the model organisms.[1] The timing of this phosphorylation varies as well. In mammalian cells, the activating phosphorylation occurs after cyclin binding.[1] In yeast cells, it occurs before cyclin binding.[1] CAK activity is not regulated by known cell-cycle pathways and cyclin binding is the limiting step for CDK activation.[1]
Unlike activating phosphorylation, CDK inhibitory phosphorylation is vital for regulation of the cell cycle. Various kinases and phosphatases regulate their phosphorylation state. One of the kinases that place the tyrosine phosphate is Wee1, a kinase conserved in all eukaryotes.[1] Fission yeast also contains a second kinase Mik1 that can phosphorylate the tyrosine.[1] Vertebrates contain a different second kinase called Myt1 that is related to Wee1 but can phosphorylate both the threonine and the tyrosine.[1] Phosphatases from the Cdc25 family dephosphorylate both the threonine and the tyrosine.[1]
CDK Inhibitors
A cyclin-dependent kinase inhibitor (CKI) is a protein that interacts with a cyclin-CDK complex to block kinase activity, usually during G1 or in response to signals from the environment or from damaged DNA.[1] In animal cells, there are two major CKI families: the INK4 family and the Cip/Kip family.[1] The INK4 family proteins are strictly inhibitory and bind CDK monomers. Crystal structures of CDK6-INK4 complexes show that INK4 binding twists the CDK to distort cyclin binding and kinase activity. The Cip/Kip family proteins bind both the cyclin and the CDK of a complex and can be inhibitory or activating. Cip/Kip proteins activate cyclin D and CDK4 or CDK6 complexes by enhancing complex formation.[1]
In yeast and Drosophila, CKIs are strong inhibitors of S- and M-CDK, but do not inhibit G1/S-CDKs. During G1, high levels of CKIs prevent cell cycle events from occurring out of order, but do not prevent transition through the Start checkpoint, which is initiated through G1/S-CDKs. Once the cell cycle is initiated, phosphorylation by early G1/S-CDKs leads to destruction of CKIs, relieving inhibition on later cell cycle transitions. In mammalian cells, the CKI regulation works differently. Mammalian protein p27 (Dacapo in Drosophila) inhibits G1/S- and S-CDKs, but does not inhibits S- and M-CDKs.[1]
Suk1 or Cks
The CDKs directly involved in the regulation of the cell cycle associate with small, 9- to 13-kiloDalton proteins called Suk1 or Cks.[3] These proteins are required for CDK function, but their precise role is unknown.[3] Cks1 binds the carboxy lobe of the Cdk, and recognizes phosphorylated residues. It may help the cyclin-CDK complex with substrates that have multiple phosphorylation sites by increasing affinity for the substrate.[3]
Non-cyclin CDK Activators
Viral Cyclins
Viruses can encode proteins with sequence homology to cyclins. One much-studied example is K-cyclin (or v-cyclin) from Kaposi sarcoma herpes virus (see Kaposi’s sarcoma), which activates CDK6. Viral cyclin-CDK complexes have different substrate specificities and regulation sensitivities.[6]
CDK5 Activators
The proteins p35 and p39 activate CDK5. Although they lack cyclin sequence homology, crystals structures show that p35 folds in a similar way as the cyclins. However, activation of CDK5 does not require activation loop phosphorylation.[6]
RINGO/Speedy
Proteins with no homology to the cyclin family can be direct activators of CDKs.[7] One family of such activators is the RINGO/Speedy family,[7] which was originally discovered in Xenopus. All five members discovered so far directly activate Cdk1 and Cdk2, but the RINGO/Speedy-CDK2 complex recognizes different substrates than cyclin A-CDK2 complex.[6]
History
Leland H. Hartwell, R. Timothy Hunt, and Paul M. Nurse received the 2001 Nobel Prize in Physiology or Medicine for their complete description of cyclin and cyclin-dependent kinase mechanisms, which are central to the regulation of the cell cycle.
Medical significance
CDKs are considered a potential target for anti-cancer medication. If it is possible to selectively interrupt the cell cycle regulation in cancer cells by interfering with CDK action, the cell will die. At present, some CDK inhibitors such as Seliciclib are undergoing clinical trials. Although it was originally developed as a potential anti-cancer drug, in recent laboratory tests Seliciclib has also proven to induce apoptosis in neutrophil granulocytes, which mediate inflammation.[8] This means that novel drugs for treatment of chronic inflammation diseases such as arthritis and cystic fibrosis could be developed.
Flavopiridol (Alvocidib) is the first CDK inhibitor to be tested in clinical trials after being identified in an anti-cancer agent screen in 1992. It competes for the ATP site of the CDKs.[9]
More research is required, however, because disruption of the CDK-mediated pathway has potentially serious consequences; while CDK inhibitors seem promising, it has to be determined how side-effects can be limited so that only target cells are affected. As such diseases are currently treated with glucocorticoids, which have often serious side-effects, even a minor success would mean an improvement.
Complications of developing a CDK drug include the fact that many CDKs are not involved in the cell cycle such as transcription, viral infection, neural physiology, and glucose homeostatsis.[10]
Table 4: Cyclin-dependent kinase inhibitor drugs [10]
Drug CDKs Inhibited Flavopiridol (Alvocidib) 1, 2, 4, 6, 7, 9 Olomoucine 1, 2, 5 Roscovitine 1, 2, 5 Purvalanol 1, 2, 5 Paullones 1, 2, 5 Butryolactone 1, 2, 5 Thio/oxoflavopiridols 1 Oxindoles 2 Aminothiazoles 4 Benzocarbazoles 4 Pyrimidines 4 Seliciclib ? References
- ^ a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab Morgan, David O. (2007). The Cell Cycle: Principles of Control. London: New Science Press, 1st ed.
- ^ Lee, Melanie; Nurse, Paul. (1987). "Complementation used to clone a human homologue of of the fission yeast cell cycle control gene cdc2." Nature 327:31-35.
- ^ a b c d e f g Morgan, David O. (1997) "Cyclin-Dependent Kinase: Engines, Clocks, and Microprocessors." Annual Review of Cell and Developmental Biology. 13:261-291.
- ^ a b Satyanarayana, A; Kaldis. (2009). “Mammalian cell-cycle regulation: several Cdks, numerous cyclins, and diverse compensatory mechanisms” “Oncogene” 28:2925-2939
- ^ Morgan, David O. (1995). “Principles of CDK regulation.” “Nature” 374:131-133.
- ^ a b c Nebreda, Angel R. (2006) “CDK activation by non-cyclin proteins.” “Current Opinion in Cell Biology.” 18:192-198
- ^ a b Mouron, Silvana; de Carcer, Guillermo; Seco, Esther; Fernandez-Miranda, Gonzalo; Malumbres, Marcos; Nebreda, Angel. (2010). "RINGO C is required to sustain the spindle assembly checkpoint." Journal of Cell Science. 123:2586-2595.
- ^ Rossi, Adriano G.; Sawatzky, Deborah A.; Walker, Annemieke; Ward, Carol; Sheldrake, Tara A.; Riley, Nicola A.; Caldicott, Alison; Martinez-Losa, Magdalena; Walker, Trevor R.; Duffin, Roger; Gray, Mohini; Crescenzi, Elvira; Martin, Morag C.; Brady, Hugh J; Savill, John S.; Dransfield, Ian & Haslett, Christopher (2006): Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nature Medicine 12 (in print). doi:10.1038/nm1468
- ^ Senderowicz, AM. “Flavopiridol: the first cyclin-dependent kinase inhibitor in human clinical trials” “Invest New Drugs” 17(3):313-20
- ^ a b Sausville, Edward A. (2002) “Complexities in the development of cyclin-dependent kinase inhibitor drugs” “Trends in Molecular Medicine” 8:S32-S37
External links
- MeSH Cyclin-Dependent+Kinases
- EC 2.7.11.22
- KEGG - Human Cell Cycle
- Loyer P, Trembley J, Katona R, Kidd V, Lahti J (2005). "Role of CDK/cyclin complexes in transcription and RNA splicing". Cell Signal 17 (9): 1033–51. doi:10.1016/j.cellsig.2005.02.005. PMID 15935619.
Cell cycle proteins Cyclin CDK CDK inhibitor P53 p63 p73 family Phases and
checkpointsOther cellular phasesCategories:
Wikimedia Foundation. 2010.