- Glucocorticoid
-
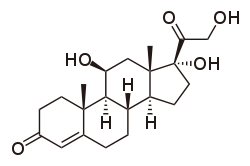 Chemical structure of cortisol, a glucocorticoid
Chemical structure of cortisol, a glucocorticoid
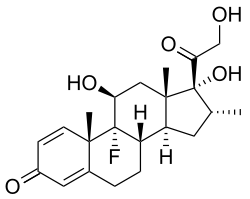 Dexamethasone binds more powerfully to the glucocorticoid receptor than cortisol does. Dexamethasone is based on the cortisol structure but differs in three positions (extra double bond in the A-ring between carbons 1 and 2 and addition of a 9-α-fluoro group and a 16-α-methyl substituent).
Dexamethasone binds more powerfully to the glucocorticoid receptor than cortisol does. Dexamethasone is based on the cortisol structure but differs in three positions (extra double bond in the A-ring between carbons 1 and 2 and addition of a 9-α-fluoro group and a 16-α-methyl substituent).Glucocorticoids (GC) are a class of steroid hormones that bind to the glucocorticoid receptor (GR), which is present in almost every vertebrate animal cell. The name glucocorticoid (glucose + cortex + steroid) derives from their role in the regulation of the metabolism of glucose, their synthesis in the adrenal cortex, and their steroidal structure (see structure to the right).
GCs are part of the feedback mechanism in the immune system that turns immune activity (inflammation) down. They are therefore used in medicine to treat diseases that are caused by an overactive immune system, such as allergies, asthma, autoimmune diseases and sepsis. GCs have many diverse (pleiotropic) effects, including potentially harmful side effects, and as a result are rarely sold over-the-counter.[1] They also interfere with some of the abnormal mechanisms in cancer cells, so they are used in high doses to treat cancer.
GCs cause their effects by binding to the glucocorticoid receptor (GR). The activated GR complex in turn up-regulates the expression of anti-inflammatory proteins in the nucleus (a process known as transactivation) and represses the expression of pro-inflammatory proteins in the cytosol by preventing the translocation of other transcription factors from the cytosol into the nucleus (transrepression).[1]
Glucocorticoids are distinguished from mineralocorticoids and sex steroids by their specific receptors, target cells, and effects. In technical terms, corticosteroid refers to both glucocorticoids and mineralocorticoids (as both are mimics of hormones produced by the adrenal cortex), but is often used as a synonym for glucocorticoid.
Cortisol (or hydrocortisone) is the most important human glucocorticoid. It is essential for life, and it regulates or supports a variety of important cardiovascular, metabolic, immunologic, and homeostatic functions. Various synthetic glucocorticoids are available; these are used either as replacement therapy in glucocorticoid deficiency or to suppress the immune system.
Contents
Effects
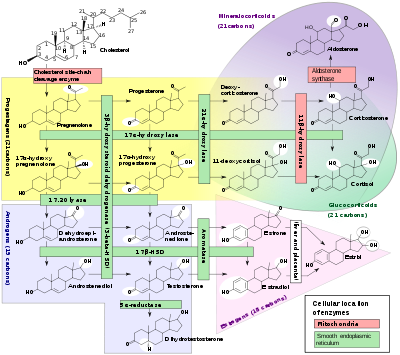 Steroidogenesis showing glucocorticoids in green ellipse at right. Note that it is not a strictly bounded group but a continuum of structures with increasing glucocorticoid effect, with the primary example being cortisol.
Steroidogenesis showing glucocorticoids in green ellipse at right. Note that it is not a strictly bounded group but a continuum of structures with increasing glucocorticoid effect, with the primary example being cortisol.Glucocorticoid effects may be broadly classified into two major categories: immunological and metabolic. In addition, glucocorticoids play important roles in fetal development.
Immune
As discussed in more detail below, glucocorticoids function through interaction with the glucocorticoid receptor:
- up-regulate the expression of anti-inflammatory proteins
- down-regulate the expression of pro-inflammatory proteins
Glucocorticoids are also shown to play a role in the development and homeostasis of T lymphocytes. This has been shown in the transgenic mice with either increased or decreased sensitivity of T cell lineage to glucocorticoids.[2]
Metabolic
The name "glucocorticoid" derives from early observations that these hormones were involved in glucose metabolism. In the fasted state, cortisol stimulates several processes that collectively serve to increase and maintain normal concentrations of glucose in blood.
Metabolic effects:
- Stimulation of gluconeogenesis, in particular, in the liver: This pathway results in the synthesis of glucose from non-hexose substrates such as amino acids and glycerol from triglyceride breakdown, and is particularly important in carnivores and certain herbivores. Enhancing the expression of enzymes involved in gluconeogenesis is probably the best-known metabolic function of glucocorticoids.
- Mobilization of amino acids from extrahepatic tissues: These serve as substrates for gluconeogenesis.
- Inhibition of glucose uptake in muscle and adipose tissue: A mechanism to conserve glucose.
- Stimulation of fat breakdown in adipose tissue: The fatty acids released by lipolysis are used for production of energy in tissues like muscle, and the released glycerol provide another substrate for gluconeogenesis.
Excessive glucocorticoid levels resulting from administration as a drug or hyperadrenocorticism have effects on many systems. Some examples include inhibition of bone formation, suppression of calcium absorption (both of which can lead to osteoporosis), delayed wound healing, muscle weakness, and increased risk of infection. These observations suggest a multitude of less-dramatic physiologic roles for glucocorticoids.[2]
Developmental
Glucocorticoids have multiple effects on fetal development. An important example is their role in promoting maturation of the lung and production of the surfactant necessary for extrauterine lung function. Mice with homozygous disruptions in the corticotropin-releasing hormone gene (see below) die at birth due to pulmonary immaturity. In addition, glucocorticoids are necessary for normal brain development, by initiating terminal maturation, remodeling axons and dendrites, and affecting cell survival.[3]
Arousal and cognition
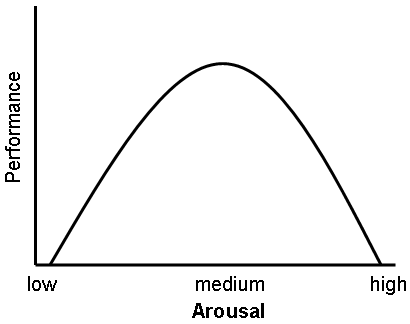
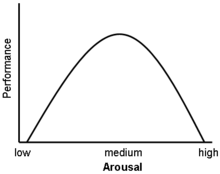 A graphical representation of the Yerkes-Dodson Curve.
A graphical representation of the Yerkes-Dodson Curve.Glucocorticoids act on the hippocampus, amygdala, and frontal lobes. Along with adrenaline, these enhance the formation of flashbulb memories of events associated with strong emotions both positive and negative.[4] This has been confirmed in studies, whereby blockade of either glucocorticoids or noradrenaline activity impaired the recall of emotionally relevant information. Additional sources have shown that subjects whose fear learning was accompanied by high cortisol levels had better consolidation of this memory (this effect was more important in men). Glucocorticoids have also been shown to have a significant impact on vigilance and cognitive performance. This appears to follow the Yerkes-Dodson Curve, as studies have shown that circulating levels of glucocorticoids vs. memory performance follow an upside-down U pattern, much like the Yerkes-Dodson curve. For example, long-term potentiation (the process of forming long-term memories) is optimal when glucocorticoid levels are mildly elevated, whereas significant decreases of LTP are observed after adrenalectomy (low-GC state) or after exogenous glucocorticoid administration (high-GC state). It has also been shown that elevated levels of glucocorticoids enhance memory for emotionally arousing events, but lead more often than not to poor memory for material unrelated to the source of stress/emotional arousal.[5] In contrast to the dose-dependent enhancing effects of glucocorticoids on memory consolidation, these stress hormones have been shown to inhibit the retrieval of already stored information.[6][7]
Mechanism of action
Transactivation
Glucocorticoids bind to the cytosolic glucocorticoid receptor (GR). This type of receptor is activated by ligand binding. After a hormone binds to the corresponding receptor, the newly formed receptor-ligand complex translocates itself into the cell nucleus, where it binds to glucocorticoid response elements (GRE) in the promoter region of the target genes resulting in the regulation of gene expression. This process is commonly referred to as transactivation.[8]
The proteins encoded by these upregulated genes have a wide range of effects including for example:[8]
- anti-inflammatory – lipocortin I, p11/calpactin binding protein and secretory leukoprotease inhibitor 1 (SLPI)
- increased gluconeogenesis – glucose-6-phosphatase and tyrosine aminotransferase
Transrepression
The opposite mechanism is called transrepression. The activated hormone receptor interacts with specific transcription factors (such as AP-1 and NF-κB) and prevents the transcription of targeted genes. Glucocorticoids are able to prevent the transcription of pro-inflammatory genes, including interleukins IL-1B, IL-4, IL-5, and IL-8, chemokines, cytokines, GM-CSF, and TNFA genes.[8]
Dissociation
The ordinary glucocorticoids do not distinguish among transactivation and transrepression and influence both the "wanted" immune and the "unwanted" genes regulating the metabolic and cardiovascular functions. Intensive research is aimed at discovering selectively acting glucocorticoids that will be able to repress only the immune system.[9][10]
Genetically modified mice that express a modified GR incapable of DNA binding are still responsive to the antiinflammatory effects of glucocorticoids, while the stimulation of gluconeogenesis by glucocorticoids is blocked.[11] This result strongly suggests that most of the desirable antiinflammatory effects are due to transrepression, while the undesirable metabolic effects arise from transactivation, a hypothesis also underlying the development of selective glucocorticoid receptor agonists.
Non-genomic
Glucocorticoids have been shown to exert a number of rapid actions that are independent of the regulations of gene transcription. Binding of corticosteroids to the glucocorticoid receptor (GR) stimulates phosphatidylinositol 3-kinase and protein kinase AKT, leading to endothelial nitric oxide synthase (eNOS) activation and nitric oxide-dependent vasorelaxation.[12] Membrane associated GR has been shown to mediate lymphocytolysis.[13][14][15] In addition, some glucocorticoids have been shown to rapidly inhibit the release of the inflammatory prostaglandin PGE2 and this effect is blocked by the glucocorticoid receptor antagonist mifepristone (RU-486) and this effect is not affected by protein synthesis inhibitors. This data together suggests a non-genomic mechanism of action.[16][17]
Glucocorticoid-induced neutrophylia
Acute or chronic administration of corticosteroids causes neutrophilia [18] suggesting that the enhanced release of PMNs from the bone marrow is an important mechanism of the glucocorticoid-induced granulocytosis. An alternative mechanism for the granulocytosis induced by glucocorticoids is an influx of PMNs from the intravascular marginated PMN pools. [19] The response is caused by a shift of cells from the marginal to the circulating pool; hence, it frequently is referred to as demargination. [20]
Some of the immunosuppressive effects of glucocorticoids are mediated by non-genomic signalling involving the glucocortiocid receptor (GR). A multi-protein complex composed of the unliganded glucocorticoid receptor, Hsp90, and the tyrosine kinases LCK and FYN is recruited to the antigen activated T cell receptor (TCR) in T cells. This GR complex is necessary for TCR signalling. On binding of glucocorticoids to GR, this multi-protein complex dissociates blocking TCR signalling.[21]
Pharmacology
A variety of synthetic glucocorticoids, some far more potent than cortisol, have been created for therapeutic use. They differ in the pharmacokinetics (absorption factor, half-life, volume of distribution, clearance) and in pharmacodynamics (for example the capacity of mineralocorticoid activity: retention of sodium (Na+) and water; see also: renal physiology). Because they permeate the intestines easily, they are administered primarily per os (by mouth), but also by other methods, such as topically on skin. More than 90 percent of them bind different plasma proteins, however with a different binding specificity. Endogenous glucocorticoids and some synthetic corticoids have high affinity to the protein transcortin (also called CBG, corticosteroid-binding globulin), whereas all of them bind albumin. In the liver, they quickly metabolise by conjugation with a sulfate or glucuronic acid, and are secreted in the urine.
Glucocorticoid potency, duration of effect, and overlapping mineralocorticoid potency varies. Cortisol (hydrocortisone) is the standard of comparison for glucocorticoid potency. Hydrocortisone is the name used for pharmaceutical preparations of cortisol. Data refer to oral dosing, except when mentioned. Oral potency may be less than parenteral potency because significant amounts (up to 50% in some cases) may not be absorbed from the intestine. Fludrocortisone, DOCA (Deoxycorticosterone acetate), and aldosterone are, by definition, not considered glucocorticoids, although they may have minor glucocorticoid potency, and are included in this table to provide perspective on mineralocorticoid potency.
Comparative steroid potencies [22] [23] Name Glucocorticoid potency Mineralocorticoid potency Duration of action (t1/2 in hours) Hydrocortisone (Cortisol) 1 1 8 Cortisone acetate 0.8 0.8 oral 8, intramuscular 18+ Prednisone 3.5-5 0.8 16-36 Prednisolone 4 0.8 16-36 Methylprednisolone 5-7.5 0.5 18-40 Dexamethasone 25-80 0 36-54 Betamethasone 25-30 0 36-54 Triamcinolone 5 0 12-36 Beclometasone 8 puffs 4 times a day
equals 14 mg oral
prednisone once a day- - Fludrocortisone acetate 15 200 24 Deoxycorticosterone acetate (DOCA) 0 20 - Aldosterone 0.3 200-1000 - Therapeutic use
Glucocorticoids may be used in low doses in adrenal insufficiency. In much higher doses, oral or inhaled glucocorticoids are used to suppress various allergic, inflammatory, and autoimmune disorders. Inhaled glucocorticoids are the second-line treatment for asthma. They are also administered as posttransplantory immunosuppressants to prevent the acute transplant rejection and the graft-versus-host disease. Nevertheless, they do not prevent an infection and also inhibit later reparative processes.
Physiological replacement
Any glucocorticoid can be given in a dose that provides approximately the same glucocorticoid effects as normal cortisol production; this is referred to as physiologic, replacement, or maintenance dosing. This is approximately 6–12 mg/m²/day (m² refers to body surface area (BSA), and is a measure of body size; an average man is 1.7 m²).
Immunosuppression
Glucocorticoids suppress the cell-mediated immunity. They act by inhibiting genes that code for the cytokines IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-8 and IFN-γ, the most important of which is IL-2. Smaller cytokine production reduces the T cell proliferation.[24]
Glucocorticoids do, however, not only reduce T cell proliferation, but also lead to another well known effect called glucocorticoid induced apoptosis. The effect is more prominent in immature T cells that still reside in the thymus, but also affect peripheral T cells. The exact mechanism underlying this glucocorticoid sensitivity still remains to be elucidated.[citation needed]
Glucocorticoids also suppress the humoral immunity, causing B cells to express smaller amounts of IL-2 and of IL-2 receptors. This diminishes both B cell clone expansion and antibody synthesis. The diminished amounts of IL-2 also causes fewer T lymphocyte cells to be activated.
Since glucocorticoid is a steroid, it regulates transcription factors; another factor it down-regulates is the expression of Fc receptors on macrophages, so there is a decreased phagocytosis of opsonised cells.[citation needed]
Anti-inflammatory
Glucocorticoids are potent anti-inflammatories, regardless of the inflammation's cause. Glucocorticoids' primary anti-inflammatory mechanism is lipocortin-1 (annexin-1) synthesis. Lipocortin-1 both suppresses phospholipase A2, thereby blocking eicosanoid production, and inhibits various leukocyte inflammatory events (epithelial adhesion, emigration, chemotaxis, phagocytosis, respiratory burst, etc.). In other words, Glucocorticoids not only suppress immune response, but also inhibit the two main products of inflammation, prostaglandins and leukotrienes. Glucocorticoids inhibit prostaglandin synthesis at the level of phospholipase A2 as well as at the level of cyclooxygenase/PGE isomerase (COX-1 and COX-2),[25] the latter effect being much like that of NSAIDs, potentiating the anti-inflammatory effect.
In addition, glucocorticoids also suppress cyclooxygenase expression.
Glucocorticoids marketed as anti-inflammatories are often topical formulations, such as nasal sprays for rhinitis or inhalers for asthma. These preparations have the advantage of only affecting the targeted area, thereby reducing side effects or potential interactions. In this case, the main compounds used are beclometasone, budesonide, fluticasone, mometasone and ciclesonide. In rhinitis, sprays are used. For asthma, glucocorticoids are administered as inhalants with a metered-dose or dry powder inhaler.[26]
Hyperaldosteronism
Glucocorticoids can be used in the management of familial hyperaldosteronism type 1. They are not effective however, for use in the type 2 condition.
Resistance
Resistance to the therapeutic uses of glucocorticoids can present difficulty; for instance, 25% of cases of severe asthma may be unresponsive to steroids. This may be the result of genetic predisposition, ongoing exposure to the cause of the inflammation (such as allergens), immunological phenomena that bypass glucocorticoids, and pharmacokinetic disturbances (incomplete absorption or accelerated excretion or metabolism).[24]
Side-effects
Glucocorticoid drugs currently being used act nonselectively, so in the long run they may impair many healthy anabolic processes. To prevent this, much research has been focused recently on the elaboration of selectively acting glucocorticoid drugs. These are the side-effects that could be prevented:
- immunosuppression
- hyperglycemia due to increased gluconeogenesis, insulin resistance, and impaired glucose tolerance ("steroid diabetes"); caution in those with diabetes mellitus
- increased skin fragility, easy bruising
- negative calcium balance due to reduced intestinal calcium absorption[27]
- Steroid-induced osteoporosis: reduced bone density (osteoporosis, osteonecrosis, higher fracture risk, slower fracture repair)
- weight gain due to increased visceral and truncal fat deposition (central obesity) and appetite stimulation
- adrenal insufficiency (if used for long time and stopped suddenly without a taper)
- muscle breakdown (proteolysis), weakness; reduced muscle mass and repair
- expansion of malar fat pads and dilation of small blood vessels in skin
- anovulation, irregularity of menstrual periods
- growth failure, pubertal delay
- increased plasma amino acids, increased urea formation; negative nitrogen balance
- excitatory effect on central nervous system (euphoria, psychosis)
- glaucoma due to increased cranial pressure
- cataracts
In high doses, hydrocortisone (cortisol) and those glucocorticoids with appreciable mineralocorticoid potency can exert a mineralocorticoid effect as well, although in physiologic doses this is prevented by rapid degradation of cortisol by 11β-hydroxysteroid dehydrogenase isoenzyme 2 (11β-HSD2) in mineralocorticoid target tissues. Mineralocorticoid effects can include salt and water retention, extracellular fluid volume expansion, hypertension, potassium depletion, and metabolic alkalosis.
The combination of clinical problems produced by prolonged, excess glucocorticoids, whether synthetic or endogenous, is termed Cushing's syndrome.
Withdrawal
In addition to the effects listed above, use of high-dose steroids for more than a week begins to produce suppression of the patient's adrenal glands because the exogenous glucocorticoids suppress hypothalamic corticotropin-releasing hormone (CRH) and pituitary adrenocorticotropic hormone (ACTH). With prolonged suppression, the adrenal glands atrophy (physically shrink), and can take months to recover full function after discontinuation of the exogenous glucocorticoid.
During this recovery time, the patient is vulnerable to adrenal insufficiency during times of stress, such as illness. While there is wide individual variation in suppressive dose and time for adrenal recovery, clinical guidelines have been devised to estimate potential adrenal suppression and recovery, to reduce risk to the patient. The following is one example, but many variations exist or may be appropriate in individual circumstances.[citation needed]
- If a patient has been receiving daily high doses for 5 days or less, they can be abruptly stopped (or reduced to physiologic replacement if patient is adrenal-deficient). Full adrenal recovery can be assumed to occur by a week afterward.
- If high doses were used for 6–10 days, reduce to replacement dose immediately and taper over 4 more days. Adrenal recovery can be assumed to occur within 2–4 weeks of completion of steroids.
- If high doses were used for 11–30 days, cut immediately to twice replacement, and then by 25% every 4 days. Stop entirely when dose is less than half of replacement. Full adrenal recovery should occur within 1–3 months of completion of withdrawal.
- If high doses were used more than 30 days, cut dose immediately to twice replacement, and reduce by 25% each week until replacement is reached.
- Then change to oral hydrocortisone or cortisone as a single morning dose, and gradually decrease by 2.5 mg each week. When a.m. dose is less than replacement, the return of normal basal adrenal function may be documented by checking 0800 cortisol levels prior to the morning dose; stop drugs when 0800 cortisol is 10 μg/dl. It is difficult to predict the time to full adrenal recovery after prolonged suppressive exogenous steroids; some people may take nearly a year.
- Flare-up of the underlying condition for which steroids are given may require a more gradual taper than outlined above.
Chemical Synthesis
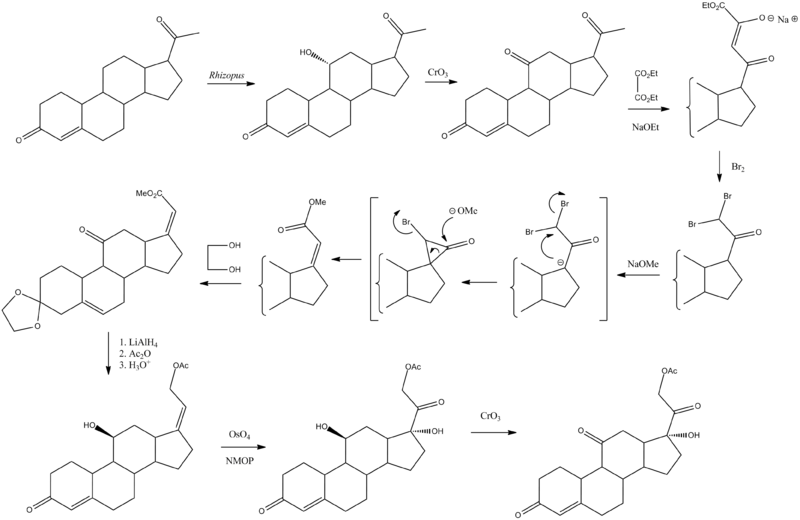
Hogg, J. A.; Beal, P. F.; Nathan, A. H.; Lincoln, F. H.; Schneider, W. P.; Magerlein, B. J.; Hanze, A. R.; Jackson, R. W. (1955). Journal of the American Chemical Society 77 (16): 4436. doi:10.1021/ja01621a092.
See also
- GITR (glucocorticoid-induced TNF receptor)
- Glucocorticoid receptor
- Immunosuppressive drug
- Selective glucocorticoid receptor agonist (SEGRA)
- Topical steroid
References
- ^ a b Rhen T, Cidlowski JA (October 2005). "Antiinflammatory action of glucocorticoids--new mechanisms for old drugs". N. Engl. J. Med. 353 (16): 1711–23. doi:10.1056/NEJMra050541. PMID 16236742.
- ^ a b Pazirandeh A, Xue Y, Prestegaard T, Jondal M, Okret S (May 2002). "Effects of altered glucocorticoid sensitivity in the T cell lineage on thymocyte and T cell homeostasis". FASEB J. 16 (7): 727–9. doi:10.1096/fj.01-0891fje. PMID 11923224.
- ^ Lupien JS, et al. (June 2009). "Effects of stress throughout the lifespan on the brain, behaviour and cognition". Nature Reviews Neuroscience 10 (6): 434–445. doi:10.1038/nrn2639. PMID 19401723.
- ^ Cahill, L., McGaugh, J. L. (1998). "Mechanisms of emotional arousal and lasting declarative memory". Trends in Neuroscience 21 (7): 294–299. doi:10.1016/S0166-2236(97)01214-9. PMID 9683321.
- ^ Lupien, S. J., Maheu, F., Tu, M.,Fiocco, A., Schramek, T.E. (2007). "The effects of stress and stress hormones on human cognition: Implications for the field of brain and cognition". Brain and Cognition 65 (3): 209–237. doi:10.1016/j.bandc.2007.02.007. PMID 17466428.
- ^ de Quervain, D et al., Stress and glucocorticoids impair retrieval of long-term spatial memory. Nature, 394, 787-790 (1998)
- ^ de Quervain, D et al., Acute cortisone administration impairs retrieval of long-term declarative memory in humans. Nature Neuroscience, 3, 313-314 (2000)
- ^ a b c Newton R (July 2000). "Molecular mechanisms of glucocorticoid action: what is important?". Thorax 55 (7): 603–13. doi:10.1136/thorax.55.7.603. PMC 1745805. PMID 10856322. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1745805.
- ^ Schäcke H, Rehwinkel H, Asadullah K (May 2005). "Dissociated glucocorticoid receptor ligands: compounds with an improved therapeutic index". Curr Opin Investig Drugs 6 (5): 503–7. PMID 15912964.
- ^ Newton R, Holden NS (October 2007). "Separating transrepression and transactivation: a distressing divorce for the glucocorticoid receptor?". Mol. Pharmacol. 72 (4): 799–809. doi:10.1124/mol.107.038794. PMID 17622575.
- ^ Reichardt HM, Tronche F, Bauer A, Schütz G (2000). "Molecular genetic analysis of glucocorticoid signaling using the Cre/loxP system". Biol. Chem. 381 (9–10): 961–4. doi:10.1515/BC.2000.118. PMID 11076028.
- ^ Hafezi-Moghadam A, Simoncini T, Yang Z, Limbourg FP, Plumier JC, Rebsamen MC, Hsieh CM, Chui DS, Thomas KL, Prorock AJ, Laubach VE, Moskowitz MA, French BA, Ley K, Liao JK (May 2002). "Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase". Nat. Med. 8 (5): 473–9. doi:10.1038/nm0502-473. PMC 2668717. PMID 11984591. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2668717.
- ^ Cato AC, Nestl A, Mink S (June 2002). "Rapid actions of steroid receptors in cellular signaling pathways". Sci. STKE 2002 (138): RE9. doi:10.1126/stke.2002.138.re9. PMID 12084906.
- ^ Gametchu B, Watson CS, Pasko D (August 1991). "Size and steroid-binding characterization of membrane-associated glucocorticoid receptor in S-49 lymphoma cells". Steroids 56 (8): 402–10. doi:10.1016/0039-128X(91)90028-T. PMID 1788858. http://linkinghub.elsevier.com/retrieve/pii/0039-128X(91)90028-T.
- ^ Gametchu B, Watson CS, Shih CC, Dashew B (August 1991). "Studies on the arrangement of glucocorticoid receptors in the plasma membrane of S-49 lymphoma cells". Steroids 56 (8): 411–9. doi:10.1016/0039-128X(91)90029-U. PMID 1788859. http://linkinghub.elsevier.com/retrieve/pii/0039-128X(91)90029-U.
- ^ Croxtall JD, van Hal PT, Choudhury Q, Gilroy DW, Flower RJ (January 2002). "Different glucocorticoids vary in their genomic and non-genomic mechanism of action in A549 cells". Br. J. Pharmacol. 135 (2): 511–9. doi:10.1038/sj.bjp.0704474. PMC 1573139. PMID 11815387. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1573139.
- ^ Revollo JR, Cidlowski JA (October 2009). "Mechanisms generating diversity in glucocorticoid receptor signaling". Ann. N. Y. Acad. Sci. 1179: 167–78. doi:10.1111/j.1749-6632.2009.04986.x. PMID 19906239.
- ^ http://emedicine.medscape.com/article/208576-overview#showall
- ^ http://circ.ahajournals.org/content/98/21/2307.full.pdf+html
- ^ Williams Hematology, 8ed, Ch.65, Neutropenia and Neutrophylia
- ^ Löwenberg M, Verhaar AP, Bilderbeek J, Marle J, Buttgereit F, Peppelenbosch MP, van Deventer SJ, Hommes DW (October 2006). "Glucocorticoids cause rapid dissociation of a T-cell-receptor-associated protein complex containing LCK and FYN". EMBO Rep. 7 (10): 1023–9. doi:10.1038/sj.embor.7400775. PMC 1618362. PMID 16888650. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1618362.
- ^ From Liapi and Chrousos (ref. 2); Chapter 14. Glucocorticoid Therapy and Adrenal Suppression; http://www.endotext.org/adrenal/adrenal14/ch01s02.html
- ^ Leung DY, Hanifin JM, Charlesworth EN, et al. (September 1997). "Disease management of atopic dermatitis: a practice parameter". Ann. Allergy Asthma Immunol. 79 (3): 197–211. doi:10.1016/S1081-1206(10)63003-7. PMID 9305225. http://www.jcaai.readyportal.net/file_depot/0-10000000/20000-30000/27387/folder/63948/Atopic_Derm1997.pdf. Retrieved 2009-07-09.
- ^ a b Leung DY, Bloom JW (January 2003). "Update on glucocorticoid action and resistance". J. Allergy Clin. Immunol. 111 (1): 3–22; quiz 23. doi:10.1067/mai.2003.97. PMID 12532089. http://www.jacionline.org/article/PIIS009167490291359X/fulltext.
- ^ Goppelt-Struebe M, Wolter D, Resch K (December 1989). "Glucocorticoids inhibit prostaglandin synthesis not only at the level of phospholipase A2 but also at the level of cyclo-oxygenase/PGE isomerase". Br. J. Pharmacol. 98 (4): 1287–95. PMC 1854794. PMID 2514948. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1854794.
- ^ Rod Flower; Humphrey P. Rang; Maureen M. Dale; Ritter, James M. (2007). Rang & Dale's pharmacology. Edinburgh: Churchill Livingstone. ISBN 0-443-06911-5.
- ^ Gennari C (May 1993). "Differential effect of glucocorticoids on calcium absorption and bone mass". Br. J. Rheumatol. 32 Suppl 2: 11–4. ISSN 1462-0332. PMID 8495275.
External links
- MeSH Glucocorticoids
- R. Bowen (2006-05-26). "Glucocorticoids". Colorado State University. http://www.vivo.colostate.edu/hbooks/pathphys/endocrine/adrenal/gluco.html. Retrieved 2008-05-11.
Corticosteroids – glucocorticoids and mineralocorticoids (H02)
(also A07EA, C05AA, D07, D10AA, R01AD, R03BA, S01BA, S02B, and S03B)Mineralocorticoids
(3-one, 4-ene,
no FG at 16)Glucocorticoids
(3-one, 4-ene,
11-FG,
17-hydroxy)PregnenePregnenedione
(+20-one)HYDROCORTISONE/CORTISOL# (Hydrocortisone aceponate, Hydrocortisone buteprate, Hydrocortisone butyrate) • Budesonide • Ciclesonide • Deflazacort • Medrysone • Tixocortol • HALOGENATED AT 6: Cloprednol • HALOGENATED, WITH FG AT 16: HalcinonidePregnadiene (+1-ene)Rimexolone • HALOGENATED, WITH FG AT 16: Flunisolide • Triamcinolone • Amcinonide • Fluocinolone acetonide (Fluocinonide)Pregnadienediol
(+21-hydroxy)Prednisone (Meprednisone) • HALOGENATED AT 9: Fluorometholone • HALOGENATED, WITH FG AT 16: Fluocortolone (Clocortolone, Diflucortolone, Fluocortin) • DesoximetasonePregnadienetriol
(+11-hydroxy)Prednisolone# (Methylprednisolone, Methylprednisolone aceponate, Prednicarbate, Prednylidene) • Desonide • HALOGENATED: Fluprednisolone (Difluprednate, Fluperolone) • HALOGENATED, WITH FG AT 16: Dexamethasone# • Betamethasone (Clobetasol, Clobetasone, Diflorasone, Halometasone, Ulobetasol) • Beclometasone • Paramethasone • Alclometasone • Fluclorolone • Flumetasone • FluprednidenePregnatriene
(+2-ene)HALOGENATED, WITH FG AT 16: Fluticasone (Fluticasone propionate, Fluticasone furoate)Other/ungroupedHALOGENATED: Loteprednol • HALOGENATED, WITH FG AT 16: Fludroxycortide • Formocortal • Mometasone furoateAldosterone antagonists Synthesis modifiers Categories:- Glucocorticoids



Wikimedia Foundation. 2010.