- Long-term potentiation
-
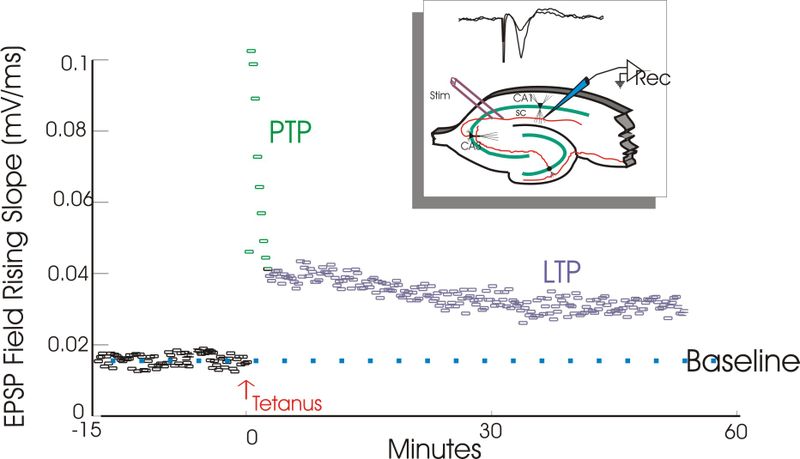
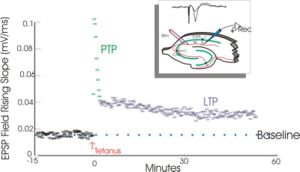 Long-term potentiation (LTP) is a persistent increase in synaptic strength following high-frequency stimulation of a chemical synapse. Studies of LTP are often carried out in slices of the hippocampus, an important organ for learning and memory. In such studies, electrical recordings are made from cells and plotted in a graph such as this one. This graph compares the response to stimuli in synapses that have undergone LTP versus synapses that have not undergone LTP. Synapses that have undergone LTP tend to have stronger electrical responses to stimuli than other synapses. The term long-term potentiation comes from the fact that this increase in synaptic strength, or potentiation, lasts a very long time compared to other processes that affect synaptic strength.[1]
Long-term potentiation (LTP) is a persistent increase in synaptic strength following high-frequency stimulation of a chemical synapse. Studies of LTP are often carried out in slices of the hippocampus, an important organ for learning and memory. In such studies, electrical recordings are made from cells and plotted in a graph such as this one. This graph compares the response to stimuli in synapses that have undergone LTP versus synapses that have not undergone LTP. Synapses that have undergone LTP tend to have stronger electrical responses to stimuli than other synapses. The term long-term potentiation comes from the fact that this increase in synaptic strength, or potentiation, lasts a very long time compared to other processes that affect synaptic strength.[1]
In neuroscience, long-term potentiation (LTP) is a long-lasting enhancement in signal transmission between two neurons that results from stimulating them synchronously.[2] It is one of several phenomena underlying synaptic plasticity, the ability of chemical synapses to change their strength. As memories are thought to be encoded by modification of synaptic strength,[3] LTP is widely considered one of the major cellular mechanisms that underlies learning and memory.[2][3]
LTP shares many features with long-term memory, making it an attractive candidate for a cellular mechanism of learning. For example, LTP and long-term memory are triggered rapidly, each depends upon the synthesis of new proteins, each has properties of associativity, and each can last for many months.[2] LTP may account for many types of learning, from the relatively simple classical conditioning present in all animals, to the more complex, higher-level cognition observed in humans.[2]
At a cellular level, LTP enhances synaptic transmission. It improves the ability of two neurons, one presynaptic and the other postsynaptic, to communicate with one another across a synapse. The precise molecular mechanisms for this enhancement of transmission have not been fully established, in part because LTP is governed by multiple mechanisms that vary by species and brain region. In the most well understood form of LTP, enhanced communication is predominantly carried out by improving the postsynaptic cell's sensitivity to signals received from the presynaptic cell.[4] These signals, in the form of neurotransmitter molecules, are received by neurotransmitter receptors present on the surface of the postsynaptic cell. LTP improves the postsynaptic cell's sensitivity to neurotransmitter in large part by increasing the activity of existing receptors and by increasing the number of receptors on the postsynaptic cell surface.[4]
LTP was discovered in the rabbit hippocampus by Terje Lømo in 1966 and has remained a popular subject of research since. Many modern LTP studies seek to better understand its basic biology, while others aim to draw a causal link between LTP and behavioral learning. Still others try to develop methods, pharmacologic or otherwise, of enhancing LTP to improve learning and memory. LTP is also a subject of clinical research, for example, in the areas of Alzheimer's disease and addiction medicine.
Contents
History
Early theories of learning
 The 19th century neuroanatomist Santiago Ramón y Cajal proposed that memories might be stored across synapses, the junctions between neurons that allow for their communication.
The 19th century neuroanatomist Santiago Ramón y Cajal proposed that memories might be stored across synapses, the junctions between neurons that allow for their communication.At the end of the 19th century, scientists generally recognized that the number of neurons in the adult brain (roughly 100 billion[5]) did not increase significantly with age, giving neurobiologists good reason to believe that memories were generally not the result of new neuron production.[6] With this realization came the need to explain how memories could form in the absence of new neurons.
The Spanish neuroanatomist Santiago Ramón y Cajal was among the first to suggest a mechanism of learning that did not require the formation of new neurons. In his 1894 Croonian Lecture, he proposed that memories might instead be formed by strengthening the connections between existing neurons to improve the effectiveness of their communication.[6] Hebbian theory, introduced by Donald Hebb in 1949, echoed Ramón y Cajal's ideas, further proposing that cells may grow new connections or undergo metabolic changes that enhance their ability to communicate:
Let us assume that the persistence or repetition of a reverberatory activity (or "trace") tends to induce lasting cellular changes that add to its stability.... When an axon of cell A is near enough to excite a cell B and repeatedly or persistently takes part in firing it, some growth process or metabolic change takes place in one or both cells such that A's efficiency, as one of the cells firing B, is increased.[7]Though these theories of memory formation are now well established, they were farsighted for their time: late 19th and early 20th century neuroscientists and psychologists were not equipped with the neurophysiological techniques necessary for elucidating the biological underpinnings of learning in animals. These skills would not come until the latter half of the 20th century, at about the same time as the discovery of long-term potentiation.
Discovery
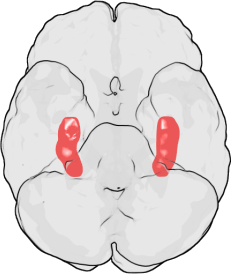
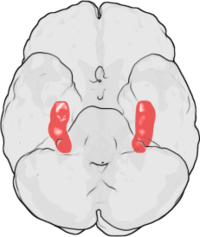 LTP was first discovered in the rabbit hippocampus. In humans, the hippocampus is located in the medial temporal lobe. This illustration of the underside of the human brain shows the hippocampus highlighted in red. The frontal lobe is at the top of the illustration and the occipital lobe is at the bottom.
LTP was first discovered in the rabbit hippocampus. In humans, the hippocampus is located in the medial temporal lobe. This illustration of the underside of the human brain shows the hippocampus highlighted in red. The frontal lobe is at the top of the illustration and the occipital lobe is at the bottom.LTP was first observed by Terje Lømo in 1966 in the Oslo, Norway, laboratory of Per Andersen.[8][9] There, Lømo conducted a series of neurophysiological experiments on anesthetized rabbits to explore the role of the hippocampus in short-term memory.
Lømo's experiments focused on connections, or synapses, from the perforant pathway to the dentate gyrus. These experiments were carried out by stimulating presynaptic fibers of the perforant pathway and recording responses from a collection of postsynaptic cells of the dentate gyrus. As expected, a single pulse of electrical stimulation to fibers of the perforant pathway caused excitatory postsynaptic potentials (EPSPs) in cells of the dentate gyrus. What Lømo unexpectedly observed was that the postsynaptic cells' response to these single-pulse stimuli could be enhanced for a long period of time if he first delivered a high-frequency train of stimuli to the presynaptic fibers. When such a train of stimuli was applied, subsequent single-pulse stimuli elicited stronger, prolonged EPSPs in the postsynaptic cell population. This phenomenon, whereby a high-frequency stimulus could produce a long-lived enhancement in the postsynaptic cells' response to subsequent single-pulse stimuli, was initially called "long-lasting potentiation".[10][11]
Timothy Bliss, who joined the Andersen laboratory in 1968,[8] collaborated with Lømo and in 1973 the two published the first characterization of long-lasting potentiation in the rabbit hippocampus.[10] Bliss and Tony Gardner-Medwin published a similar report of long-lasting potentiation in the awake animal which appeared in the same issue as the Bliss and Lømo report.[11] In 1975, Douglas and Goddard proposed "long-term potentiation" as a new name for the phenomenon of long-lasting potentiation.[12][13] Andersen suggested that the authors chose "long-term potentiation" perhaps because of its easily pronounced acronym, "LTP".[14]
Models and theory
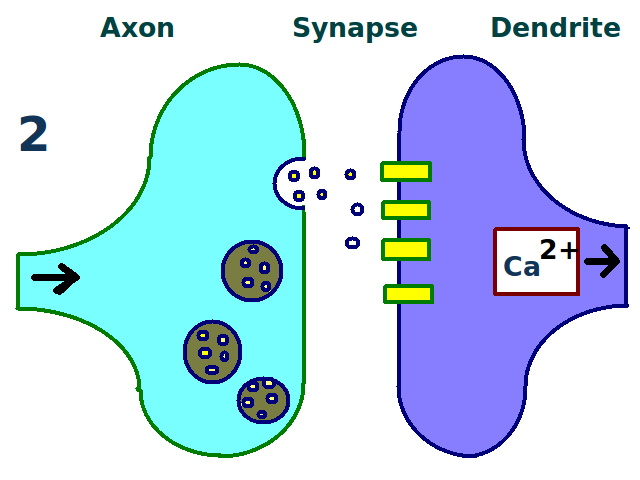
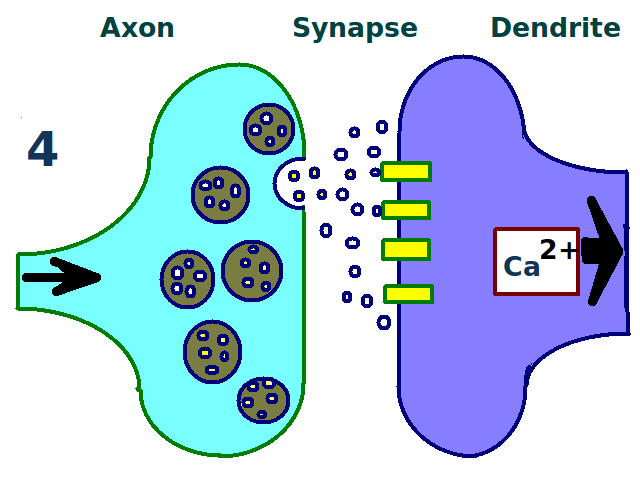
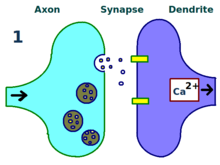 A synapse is repeatedly stimulated.
A synapse is repeatedly stimulated.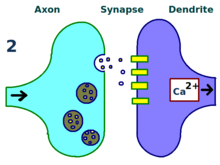 More dendritic receptors.
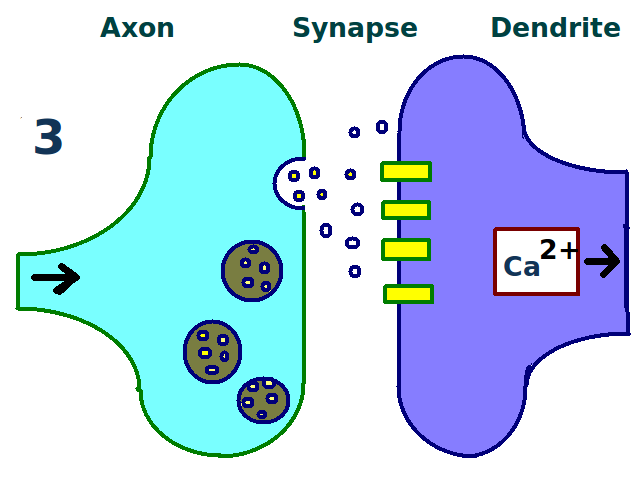
More dendritic receptors.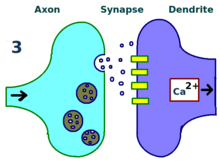 More neurotransmitters.
More neurotransmitters.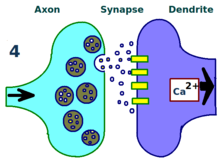 A stronger link between neurons.
A stronger link between neurons.The physical and biological mechanism of LTP is still not understood, but some successful models have been developed.[1] Studies of dendritic spines, protruding structures on dendrites that physically grow and retract over the course of minutes or hours, have suggested a relationship between the electrical resistance of the spine and the effective synapse strength, due to their relationship with intracellular calcium transients. Mathematical models such as BCM Theory, which depends also on intracellular calcium in relation to NMDA receptor voltage gates, have been developed since the 1980s and modify the traditional a priori Hebbian learning model with both biological and experimental justification. Still others have proposed re-arranging or synchronizing the relationship between receptor regulation, LTP, and synaptic strength.[15]
Types
Since its original discovery in the rabbit hippocampus, LTP has been observed in a variety of other neural structures, including the cerebral cortex, cerebellum, amygdala,[16] and many others. Robert Malenka, a prominent LTP researcher, has suggested that LTP may even occur at all excitatory synapses in the mammalian brain.[4]
Different areas of the brain exhibit different forms of LTP. The specific type of LTP exhibited between neurons depends on a number of factors. One such factor is the age of the organism when LTP is observed. For example, the molecular mechanisms of LTP in the immature hippocampus differ from those mechanisms that underlie LTP of the adult hippocampus.[17] The signalling pathways used by a particular cell also contribute to the specific type of LTP present. For example, some types of hippocampal LTP depend on the NMDA receptor, others may depend upon the metabotropic glutamate receptor (mGluR), while still others depend upon another molecule altogether.[4] The variety of signaling pathways that contribute to LTP and the wide distribution of these various pathways in the brain are reasons that the type of LTP exhibited between neurons depends in part upon the anatomic location in which LTP is observed. For example, LTP in the Schaffer collateral pathway of the hippocampus is NMDA receptor-dependent, whereas LTP in the mossy fiber pathway is NMDA receptor-independent.[18]
The pre- and postsynaptic activity required to induce LTP are other criteria by which LTP is classified. Broadly, this allows classification of LTP into Hebbian, non-Hebbian, and anti-Hebbian mechanisms. Borrowing its name from Hebb's postulate, summarized by the maxim that "cells that fire together wire together," Hebbian LTP requires simultaneous pre- and postsynaptic depolarization for its induction.[19] Non-Hebbian LTP is a type of LTP that does not require such simultaneous depolarization of pre- and postsynaptic cells; an example of this occurs in the mossy fiber hippocampal pathway.[20] A special case of non-Hebbian LTP, anti-Hebbian LTP explicitly requires simultaneous presynaptic depolarization and relative postsynaptic hyperpolarization for its induction.[21]
Owing to its predictable organization and readily inducible LTP, the CA1 hippocampus has become the prototypical site of mammalian LTP study. In particular, NMDA receptor-dependent LTP in the adult CA1 hippocampus is the most widely studied type of LTP,[4] and is therefore the focus of this article.
Properties
NMDA receptor-dependent LTP exhibits several properties, including input specificity, associativity, cooperativity, and persistence.
- Input specificity
- Once induced, LTP at one synapse does not spread to other synapses; rather LTP is input specific. Long-term potentiation is only propagated to those synapses according to the rules of associativity and cooperativity. However, the input specificity of LTP may be incomplete at short distances. One model to explain the input specificity of LTP was presented by Frey and Morris in 1997 and is called the synaptic tagging and capture hypothesis.
- Associativity
- Associativity refers to the observation that when weak stimulation of a single pathway is insufficient for the induction of LTP, simultaneous strong stimulation of another pathway will induce LTP at both pathways.
- Cooperativity
- LTP can be induced either by strong tetanic stimulation of a single pathway to a synapse, or cooperatively via the weaker stimulation of many. When one pathway into a synapse is stimulated weakly, it produces insufficient postsynaptic depolarization to induce LTP. In contrast, when weak stimuli are applied to many pathways that converge on a single patch of postsynaptic membrane, the individual postsynaptic depolarizations generated may collectively depolarize the postsynaptic cell enough to induce LTP cooperatively. Synaptic tagging, discussed later, may be a common mechanism underlying associativity and cooperativity. Bruce McNaughton argues that any difference between associativity and cooperativity is strictly semantic.[22]
- Persistence
- LTP is persistent, lasting from several minutes to many months, and it is this persistence that separates LTP from other forms of synaptic plasticity.[23]
Mechanism
Long-term potentiation occurs through a variety of mechanisms throughout the nervous system; no single mechanism unites all of LTP's many types. However, for the purposes of study, LTP is commonly divided into three phases that occur sequentially: short-term potentiation, early LTP, and late LTP.[24] Little is known about the mechanisms of short-term potentiation,[24] so it will not be discussed here.
Each phase of LTP is governed by a set of mediators, small molecules that dictate the events of that phase.[4] These molecules include protein receptors that respond to events outside of the cell, enzymes that carry out chemical reactions within the cell, and signaling molecules that allow the progression from one phase to the next. In addition to these mediators, there are also modulator molecules, described later, that interact with mediators to finely alter the LTP ultimately generated.
The early (E-LTP) and late (L-LTP) phases of LTP are each characterized by a series of three events: induction, maintenance, and expression. Induction is the process by which a short-lived signal triggers that phase of LTP to begin. Maintenance corresponds to the persistent biochemical changes that occur in response to the induction of that phase. Expression entails the long-lasting cellular changes that result from activation of the maintenance signal.[24] Thus the mechanisms of LTP can be discussed in terms of the mediators that underlie the induction, maintenance, and expression of E-LTP and L-LTP.
Early phase

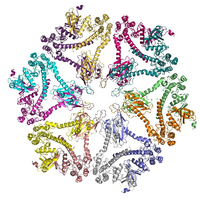 Ca2+/calmodulin-dependent protein kinase II (CaMKII) appears to be an important mediator of the early, protein synthesis-independent phase of LTP.
Ca2+/calmodulin-dependent protein kinase II (CaMKII) appears to be an important mediator of the early, protein synthesis-independent phase of LTP.Induction
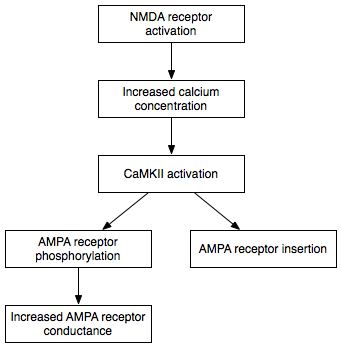
Induction of E-LTP occurs when the concentration of calcium inside the postsynaptic cell exceeds a critical threshold.[25] In many types of LTP, the flow of calcium into the cell requires the NMDA receptor, which is why these types of LTP are considered to be NMDA receptor-dependent.[25] NMDA receptor-dependent LTP can be induced experimentally by applying a few trains of high-frequency stimuli to the connection between two neurons.[26] An understanding of normal synaptic transmission illustrates how this tetanic stimulation can induce E-LTP.
Chemical synapses are functional connections between neurons throughout the nervous system. In a typical synapse, information is passed from the first (presynaptic) neuron to the second (postsynaptic) neuron via a process of synaptic transmission. Through experimental manipulation, a non-tetanic stimulus can be applied to the presynaptic cell, causing it to release a neurotransmitter—typically glutamate—onto the postsynaptic cell membrane. There, glutamate binds to AMPA receptors (AMPARs) embedded in the postsynaptic membrane. The AMPA receptor is one of the main excitatory receptors in the brain, and is responsible for most of its rapid, moment-to-moment excitatory activity.[27] Glutamate binding to the AMPA receptor triggers the influx of positively charged sodium ions into the postsynaptic cell, causing a short-lived depolarization called the excitatory postsynaptic potential (EPSP).
The magnitude of this depolarization determines whether E-LTP will be induced in the postsynaptic cell. While a single stimulus does not generate an EPSP capable of inducing E-LTP, repeated stimuli given at high frequency cause the postsynaptic cell to be progressively depolarized as a result of EPSP summation: with each EPSP reaching the postsynaptic cell before the previous EPSP can decay, successive EPSPs add to the depolarization caused by the previous EPSPs. In synapses that exhibit NMDA receptor-dependent LTP, sufficient depolarization unblocks NMDA receptors (NMDARs), receptors that allow calcium to flow into the cell when bound by glutamate. While NMDARs are present at most postsynaptic membranes, at resting membrane potentials they are blocked by a magnesium ion that prevents the entry of calcium into the postsynaptic cell. Sufficient depolarization through the summation of EPSPs relieves the magnesium blockade of the NMDAR, allowing calcium influx (despite the reduced driving force for calcium entry). The rapid rise in intracellular calcium concentration triggers the short-lasting activation of several enzymes that mediate E-LTP induction. Of particular importance are some protein kinase enzymes, including calcium/calmodulin-dependent protein kinase II (CaMKII) and protein kinase C (PKC).[24] To a lesser extent, protein kinase A (PKA) and mitogen-activated protein kinase (MAPK) activation also contribute to the induction of E-LTP.[24]
Maintenance
While induction entails the transient activation of CaMKII and PKC, maintenance of E-LTP is characterized by their persistent activation. During this stage, PKMz(Protein kinase Mζ) which does not have dependence on calcium, become autonomously active. Consequently they are able to carry out the phosphorylation events that underlie E-LTP expression.[24]
Expression
Phosphorylation is a chemical reaction in which a small phosphate group is added to another molecule to change that molecule's activity. Autonomously active CaMKII and PKC use phosphorylation to carry out the two major mechanisms underlying the expression of E-LTP. First, and most importantly, they phosphorylate existing AMPA receptors to increase their activity.[4] Second, they mediate or modulate the insertion of additional AMPA receptors into the postsynaptic membrane.[4] Importantly, the delivery of AMPA receptors to the synapse during E-LTP is independent of protein synthesis. This is achieved by having a nonsynaptic pool of AMPA receptors adjacent to the postsynaptic membrane. When the appropriate LTP-inducing stimulus arrives, nonsynaptic AMPA receptors are rapidly trafficked into the postsynaptic membrane under the influence of protein kinases.[28] As mentioned previously, AMPA receptors are the brain's most abundant glutamate receptors and mediate the majority of its excitatory activity. By increasing the efficiency and number of AMPA receptors at the synapse, future excitatory stimuli generate larger postsynaptic responses.
While the above model of E-LTP describes entirely postsynaptic mechanisms for induction, maintenance, and expression, an additional component of expression may occur presynaptically.[29] One hypothesis of this presynaptic facilitation is that persistent CaMKII activity during E-LTP may lead to the synthesis of a "retrograde messenger", discussed later. According to this hypothesis, the newly synthesized messenger travels across the synaptic cleft from the postsynaptic to the presynaptic cell, leading to a chain of events that facilitate the presynaptic response to subsequent stimuli. Such events may include an increase in neurotransmitter vesicle number, probability of vesicle release, or both. In addition to the retrograde messenger underlying presynaptic expression in early LTP, the retrograde messenger may also play a role in the expression of late LTP.
Late phase
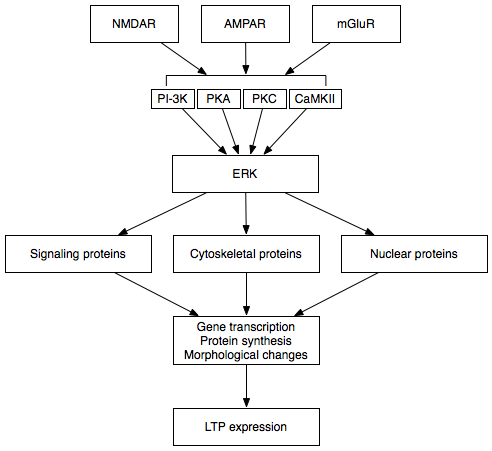
 The early and late phases of LTP are thought to communicate via the extracellular signal-regulated kinase (ERK).[25]
The early and late phases of LTP are thought to communicate via the extracellular signal-regulated kinase (ERK).[25]Late LTP is the natural extension of E-LTP. Unlike E-LTP, which is independent of protein synthesis, L-LTP requires gene transcription[30] and protein synthesis[31] in the postsynaptic cell. Two phases of L-LTP exist: the first depends upon protein synthesis, while the second depends upon both gene transcription and protein synthesis.[25] These phases are occasionally called LTP2 and LTP3, respectively, with E-LTP referred to as LTP1 under this nomenclature.
Induction
Late LTP is induced by changes in gene expression and protein synthesis brought about by the persistent activation of protein kinases activated during E-LTP, such as MAPK.[24][25][32] In fact, MAPK—specifically the extracellular signal-regulated kinase (ERK) subfamily of MAPKs—may be the molecular link between E-LTP and L-LTP, since many signaling cascades involved in E-LTP, including CaMKII and PKC, can converge on ERK.[32] Recent research has shown that the induction of L-LTP can depend on coincident molecular events, namely PKA activation and calcium influx, that converge on CRTC1 (TORC1), a potent transcriptional coactivator for cAMP response element binding protein (CREB).[33] This requirement for a molecular coincidence accounts perfectly for the associative nature of LTP, and, presumably, for that of learning.
Maintenance
Upon activation, ERK may phosphorylate a number of cytoplasmic and nuclear molecules that ultimately result in the protein synthesis and morphological changes observed in L-LTP.[25] These cytoplasmic and nuclear molecules may include transcription factors such as CREB.[24] ERK-mediated changes in transcription factor activity may trigger the synthesis of proteins that underlie the maintenance of L-LTP. One such molecule may be protein kinase Mζ (PKMζ), a persistently active kinase whose synthesis increases following LTP induction.[34][35] PKMζ is an atypical isoform of PKC that lacks a regulatory subunit and thus remains constitutively active.[34] Unlike other kinases that mediate LTP, PKMζ is active not just in the first 30 minutes following LTP induction; rather, PKMζ becomes a requirement for LTP maintenance only during the late phase of LTP.[34] PKMζ thus appears important for the persistence of memory and would be expected to be important in the maintenance of long-term memory. Indeed, administration of a PKMζ inhibitor into the hippocampi of rats results in retrograde amnesia with intact short-term memory; PKMζ does not play a role in the establishment of short-term memory.[35] PKMζ has recently been shown to underlie L-LTP maintenance[34][35] by directing the trafficking and reorganization of proteins in the synaptic scaffolding that underlie the expression of L-LTP.[34]
Expression
Aside from PKMζ, the identities of only a few proteins synthesized during L-LTP are known. Regardless of their identities, it is thought that they contribute to the increase in dendritic spine number, surface area, and postsynaptic sensitivity to neurotransmitter associated with L-LTP expression.[25] The latter may be brought about in part by the enhanced synthesis of AMPA receptors during L-LTP.[25] Late LTP is also associated with the presynaptic synthesis of synaptotagmin and an increase in synaptic vesicle number, suggesting that L-LTP induces protein synthesis not only in postsynaptic cells, but in presynaptic cells as well.[25] As mentioned previously, for postsynaptic LTP induction to result in presynaptic protein synthesis, there must be communication from the postsynaptic to the presynaptic cell. This may occur via the synthesis of a retrograde messenger, discussed later.
Even in studies restricted to postsynaptic events, investigators have not determined the location of the protein synthesis that underlies L-LTP. Specifically, it is unclear whether protein synthesis takes place in the postsynaptic cell body or in its dendrites.[32] Despite having observed ribosomes (the major components of the protein synthesis machinery) in dendrites as early as the 1960s, prevailing wisdom was that the cell body was the predominant site of protein synthesis in neurons.[32] This reasoning was not seriously challenged until the 1980s, when investigators reported observing protein synthesis in dendrites whose connection to their cell body had been severed.[32] More recently, investigators have demonstrated that this type of local protein synthesis is necessary for some types of LTP.[36][37]
One reason for the popularity of the local protein synthesis hypothesis is that it provides a possible mechanism for the specificity associated with LTP.[32] Specifically, if indeed local protein synthesis underlies L-LTP, only dendritic spines receiving LTP-inducing stimuli will undergo LTP; the potentiation will not be propagated to adjacent synapses. By contrast, global protein synthesis that occurs in the cell body requires that proteins be shipped out to every area of the cell, including synapses that have not received LTP-inducing stimuli. Whereas local protein synthesis provides a mechanism for specificity, global protein synthesis would seem to directly compromise it. However, as discussed later, the synaptic tagging hypothesis successfully reconciles global protein synthesis, synapse specificity, and associativity.
Retrograde signaling
Retrograde signaling is a hypothesis that attempts to explain that, while LTP is induced and expressed postsynaptically, some evidence suggests that it is expressed presynaptically as well.[4][29][38] The hypothesis gets its name because normal synaptic transmission is directional and proceeds from the presynaptic to the postsynaptic cell. For induction to occur postsynaptically and be partially expressed presynaptically, a message must travel from the postsynaptic cell to the presynaptic cell in a retrograde (reverse) direction. Once there, the message presumably initiates a cascade of events that leads to a presynaptic component of expression, such as the increased probability of neurotransmitter vesicle release.[39]
Retrograde signaling is currently a contentious subject as some investigators do not believe the presynaptic cell contributes at all to the expression of LTP.[4] Even among proponents of the hypothesis there is controversy over the identity of the messenger. Early thoughts focused on nitric oxide, while most recent evidence points to cell adhesion proteins.[4]
Synaptic tagging
Before the local protein synthesis hypothesis gained significant support, there was general agreement that the protein synthesis underlying L-LTP occurred in the cell body. Further, there was thought that the products of this synthesis were shipped cell-wide in a nonspecific manner. It thus became necessary to explain how protein synthesis could occur in the cell body without compromising LTP's input specificity. The synaptic tagging hypothesis attempts to solve the cell's difficult problem of synthesizing proteins in the cell body but ensuring they only reach synapses that have received LTP-inducing stimuli.
The synaptic tagging hypothesis proposes that a "synaptic tag" is synthesized at synapses that have received LTP-inducing stimuli, and that this synaptic tag may serve to capture plasticity-related proteins shipped cell-wide from the cell body.[40] Studies of LTP in the marine snail Aplysia californica have implicated synaptic tagging as a mechanism for the input-specificity of LTP.[41][42] There is some evidence that given two widely separated synapses, an LTP-inducing stimulus at one synapse drives several signaling cascades (described previously) that initiates gene expression in the cell nucleus. At the same synapse (but not the unstimulated synapse), local protein synthesis creates a short-lived (less than three hours) synaptic tag. The products of gene expression are shipped globally throughout the cell, but are only captured by synapses that express the synaptic tag. Thus only the synapse receiving LTP-inducing stimuli is potentiated, demonstrating LTP's input specificity.
The synaptic tag hypothesis may also account for LTP's associativity and cooperativity. Associativity (see Properties) is observed when one synapse is excited with LTP-inducing stimulation while a separate synapse is only weakly stimulated. Whereas one might expect only the strongly stimulated synapse to undergo LTP (since weak stimulation alone is insufficient to induce LTP at either synapse), both synapses will in fact undergo LTP. While weak stimuli are unable to induce protein synthesis in the cell body, they may prompt the synthesis of a synaptic tag. Simultaneous strong stimulation of a separate pathway, capable of inducing cell body protein synthesis, then may prompt the production of plasticity-related proteins, which are shipped cell-wide. With both synapses expressing the synaptic tag, both would capture the protein products resulting in the expression of LTP in both the strongly stimulated and weakly stimulated pathways.
Cooperativity is observed when two synapses are activated by weak stimuli incapable of inducing LTP when stimulated individually. But upon simultaneous weak stimulation, both synapses undergo LTP in a cooperative fashion. Synaptic tagging does not explain how multiple weak stimuli can result in a collective stimulus sufficient to induce LTP (this is explained by the postsynaptic summation of EPSPs described previously). Rather, synaptic tagging explains the ability of weakly stimulated synapses, none of which are capable of independently generating LTP, to receive the products of protein synthesis initiated collectively. As before, this may be accomplished through the synthesis of a local synaptic tag following weak synaptic stimulation.
Modulation
Proposed modulators of LTP[24] Modulator Target β-Adrenergic receptor cAMP, MAPK amplification Nitric oxide synthase Guanylyl cyclase, PKG, NMDAR Dopamine receptor cAMP, MAPK amplification Metabotropic glutamate receptor PKC, MAPK amplification As described previously, the molecules that underlie LTP can be classified as mediators or modulators. A mediator of LTP is a molecule, such as the NMDA receptor or calcium, whose presence and activity is necessary for generating LTP under nearly all conditions. By contrast, a modulator is a molecule that can alter LTP but is not essential for its generation or expression.[4]
In addition to the signaling pathways described above, hippocampal LTP may be altered by a variety of modulators. For example, the steroid hormone estradiol may enhance LTP by driving CREB phosphorylation and subsequent dendritic spine growth.[43] Additionally, β-adrenergic receptor agonists such as norepinephrine may alter the protein synthesis-dependent late phase of LTP.[44] Nitric oxide synthase activity may also result in the subsequent activation of guanylyl cyclase and PKG.[45] Similarly, activation of dopamine receptors may enhance LTP through the cAMP/PKA signaling pathway.[46][47]
Relationship to behavioral memory
While the long-term potentiation of synapses in cell culture seems to provide an elegant substrate for learning and memory, the contribution of LTP to behavioral learning — that is, learning at the level of the whole organism — cannot simply be extrapolated from in vitro studies. For this reason, considerable effort has been dedicated to establishing whether LTP is a requirement for learning and memory in living animals.
Spatial memory
 The Morris water maze task has been used to demonstrate the necessity of NMDA receptors in establishing spatial memories.
The Morris water maze task has been used to demonstrate the necessity of NMDA receptors in establishing spatial memories.In 1986, Richard Morris provided some of the first evidence that LTP was indeed required for the formation of memories in vivo.[48] He tested the spatial memory of rats by pharmacologically modifying their hippocampus, a brain structure whose role in spatial learning is well established. Rats were trained on the Morris water maze, a spatial memory task in which rats swim in a pool of murky water until they locate the platform hidden beneath its surface. During this exercise, normal rats are expected to associate the location of the hidden platform with salient cues placed at specific positions around the circumference of the maze. After training, one group of rats had their hippocampi bathed in the NMDA receptor blocker APV, while the other group served as the control. Both groups were then subjected to the water maze spatial memory task. Rats in the control group were able to locate the platform and escape from the pool, while the performance of APV-treated rats was significantly impaired. Moreover, when slices of the hippocampus were taken from both groups, LTP was easily induced in controls, but could not be induced in the brains of APV-treated rats. This provided early evidence that the NMDA receptor — and by extension, LTP — was required for at least some types of learning and memory.
Similarly, Susumu Tonegawa demonstrated in 1996 that the CA1 area of the hippocampus is crucial to the formation of spatial memories in living mice.[49] So-called place cells located in this region become active only when the rat is in a particular location — called a place field — in the environment. Since these place fields are distributed throughout the environment, one interpretation is that groups of place cells form maps in the hippocampus. The accuracy of these maps determines how well a rat learns about its environment and thus how well it can navigate it. Tonegawa found that by impairing the NMDA receptor, specifically by genetically removing the NR1 subunit in the CA1 region, the place fields generated were substantially less specific than those of controls. That is, rats produced faulty spatial maps when their NMDA receptors were impaired. As expected, these rats performed very poorly on spatial tasks compared to controls, further supporting the role of LTP in spatial learning.
Enhanced NMDA receptor activity in the hippocampus has also been shown to produce enhanced LTP and an overall improvement in spatial learning. In 1999, Tang et al. produced a line of mice with enhanced NMDA receptor function by overexpressing the NR2B subunit in the hippocampus.[50][51] The resulting smart mice, nicknamed "Doogie mice" after the fictional prodigious doctor Doogie Howser, had larger LTP and excelled at spatial learning tasks, reinforcing LTP's importance in the formation of hippocampus-dependent memories.
Inhibitory avoidance
In 2006, Jonathan Whitlock and colleagues reported on a series of experiments that provided perhaps the strongest evidence of LTP's role in behavioral memory, arguing that to conclude that LTP underlies behavioral learning, the two processes must both mimic and occlude one another.[52] Employing an inhibitory avoidance learning paradigm, researchers trained rats in a two-chambered apparatus with light and dark chambers, the latter being fitted with a device that delivered a foot shock to the rat upon entry. An analysis of CA1 hippocampal synapses revealed that inhibitory avoidance training induced in vivo AMPA receptor phosphorylation of the same type as that seen in LTP in vitro; that is, inhibitory avoidance training mimicked LTP. In addition, synapses potentiated during training could not be further potentiated by experimental manipulations that would have otherwise induced LTP; that is, inhibitory avoidance training occluded LTP. In a response to the article, Timothy Bliss and colleagues remarked that these and related experiments "substantially advance the case for LTP as a neural mechanism for memory."[53]
Clinical significance
The role of LTP in disease is less clear than its role in basic mechanisms of synaptic plasticity. However, alterations in LTP may contribute to a number of neurological diseases, including depression, Parkinson's disease, epilepsy, and neuropathic pain.[54] Impaired LTP may also have a role in Alzheimer's disease and drug addiction.
Alzheimer's disease
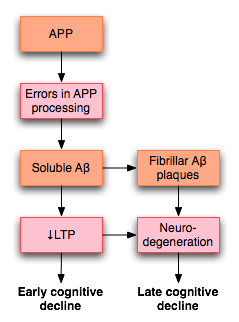
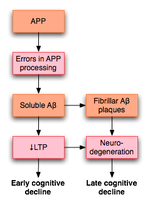 Misprocessing of amyloid precursor protein (APP) in Alzheimer's disease disrupts LTP and is thought to lead to early cognitive decline in individuals with the disease.[55]
Misprocessing of amyloid precursor protein (APP) in Alzheimer's disease disrupts LTP and is thought to lead to early cognitive decline in individuals with the disease.[55]LTP has received much attention among those who study Alzheimer's disease (AD), a neurodegenerative disease that causes marked cognitive decline and dementia. Much of this deterioration occurs in association with degenerative changes in the hippocampus and other medial temporal lobe structures. Because of the hippocampus' well established role in LTP, some have suggested that the cognitive decline seen in individuals with AD may result from impaired LTP.
In a 2003 review of the literature, Rowan et al. proposed one model for how LTP might be affected in AD.[55] AD appears to result, at least in part, from misprocessing of amyloid precursor protein (APP). The result of this abnormal processing is the accumulation of fragments of this protein, called amyloid β (Aβ). Aβ exists in both soluble and fibrillar forms. Misprocessing of APP results in the accumulation of soluble Aβ that, according to Rowan's hypothesis, impairs hippocampal LTP and may lead to the cognitive decline seen early in AD.
AD may also impair LTP through mechanisms distinct from Aβ. For example, one study demonstrated that the enzyme PKMζ accumulates in neurofibrillary tangles, which are a pathologic marker of AD. PKMζ is an enzyme with critical importance in the maintenance of late LTP.[56]
Drug addiction
Research in the field of addiction medicine has also recently turned its focus to LTP, owing to the hypothesis that drug addiction represents a powerful form of learning and memory.[57] Addiction is a complex neurobehavioral phenomenon involving various parts of the brain, such as the ventral tegmental area (VTA) and nucleus accumbens (NAc). Studies have demonstrated that VTA and NAc synapses are capable of undergoing LTP[57] and that this LTP may be responsible for the behaviors that characterize addiction.[58]
See also
- Synaptic plasticity
- Neuroplasticity
- Long-term depression
- Long-term memory
- Actin remodeling of neurons
References
- ^ Paradiso, Michael A.; Bear, Mark F.; Connors, Barry W. (2007). Neuroscience: Exploring the Brain. Hagerstwon, MD: Lippincott Williams & Wilkins. p. 718. ISBN 0-7817-6003-8.
- ^ a b c d Cooke SF, Bliss TV (2006). "Plasticity in the human central nervous system". Brain 129 (Pt 7): 1659–73. doi:10.1093/brain/awl082. PMID 16672292.
- ^ a b Bliss TV, Collingridge GL (January 1993). "A synaptic model of memory: long-term potentiation in the hippocampus". Nature 361 (6407): 31–39. doi:10.1038/361031a0. PMID 8421494.
- ^ a b c d e f g h i j k l Malenka R, Bear M (2004). "LTP and LTD: an embarrassment of riches". Neuron 44 (1): 5–21. doi:10.1016/j.neuron.2004.09.012. PMID 15450156.
- ^ Williams RW, Herrup K (1988). "The control of neuron number". Annu. Rev. Neurosci. 11 (1): 423–53. doi:10.1146/annurev.ne.11.030188.002231. PMID 3284447. http://www.nervenet.org/papers/NUMBER_REV_1988.html.
- ^ a b Ramón y Cajal, Santiago (1894). "The Croonian Lecture: La Fine Structure des Centres Nerveux". Proceedings of the Royal Society of London 55 (331-335): 444–468. doi:10.1098/rspl.1894.0063.
- ^ Hebb, D. O. (1949). Organization of Behavior: a Neuropsychological Theory. New York: John Wiley. ISBN 0-471-36727-3.
- ^ a b Terje Lømo (2003). "The discovery of long-term potentiation". Philos Trans R Soc Lond B Biol Sci 358 (1432): 617–20. doi:10.1098/rstb.2002.1226. PMC 1693150. PMID 12740104. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1693150.
- ^ Lømo, Terje (1966). "Frequency potentiation of excitatory synaptic activity in the dentate area of the hippocampal formation". Acta Physiologica Scandinavica 68 (Suppl 277): 128.
- ^ a b Bliss T, Lømo T (1973). "Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path". J Physiol 232 (2): 331–56. PMC 1350458. PMID 4727084. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1350458.
- ^ a b Bliss T, Gardner-Medwin A (1973). "Long-lasting potentiation of synaptic transmission in the dentate area of the unanaestetized rabbit following stimulation of the perforant path". J. Physiol. (Lond.) 232 (2): 357–74. PMC 1350459. PMID 4727085. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1350459.
- ^ While the term "long term potentiation" appeared once in the original Bliss and Lømo paper, it was not formally proposed for the phenomenon until the Douglas and Goddard paper.
- ^ Douglas R, Goddard G (1975). "Long-term potentiation of the perforant path-granule cell synapse in the rat hippocampus". Brain Res. 86 (2): 205–15. doi:10.1016/0006-8993(75)90697-6. PMID 163667.
- ^ Andersen P (2003). "A prelude to long-term potentiation". Philos. Trans. R. Soc. Lond., B, Biol. Sci. 358 (1432): 613–5. doi:10.1098/rstb.2002.1232. PMC 1693144. PMID 12740103. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1693144.
- ^ McEachern, JC; Shaw, CA (June 1996). "An alternative to the LTP orthodoxy: a plasticity-pathology continuum model". Brain Research Review 22 (1): 51–92. doi:10.1016/0165-0173(96)00006-9. PMID 8871785. 8871785.
- ^ Clugnet, MC; LeDoux JE (1 August 1990). "Synaptic plasticity in fear conditioning circuits: induction of LTP in the lateral nucleus of the amygdala by stimulation of the medial geniculate body." (PDF). J Neurosci 10 (8): 2818–24. PMID 2388089. http://www.jneurosci.org/cgi/reprint/10/8/2818.
- ^ Yasuda H, Barth A, Stellwagen D, Malenka R (2003). "A developmental switch in the signaling cascades for LTP induction". Nat Neurosci 6 (1): 15–6. doi:10.1038/nn985. PMID 12469130.
- ^ Harris E, Cotman C (1986). "Long-term potentiation of guinea pig mossy fiber responses is not blocked by N-methyl D-aspartate antagonists". Neurosci Lett 70 (1): 132–7. doi:10.1016/0304-3940(86)90451-9. PMID 3022192.
- ^ Wigström H, Gustafsson B (1986). "Postsynaptic control of hippocampal long-term potentiation". J. Physiol. (Paris) 81 (4): 228–36. PMID 2883309.
- ^ Urban NN, Barrionuevo G (July 1996). "Induction of hebbian and non-hebbian mossy fiber long-term potentiation by distinct patterns of high-frequency stimulation". J. Neurosci. 16 (13): 4293–9. PMID 8753890. http://www.jneurosci.org/cgi/pmidlookup?view=long&pmid=8753890.
- ^ Kullmann DM, Lamsa K (March 2008). "Roles of distinct glutamate receptors in induction of anti-Hebbian long-term potentiation". J. Physiol. (Lond.) 586 (6): 1481–6. doi:10.1113/jphysiol.2007.148064. PMC 2375711. PMID 18187472. http://www.jphysiol.org/cgi/pmidlookup?view=long&pmid=18187472.
- ^ McNaughton BL (April 2003). "Long-term potentiation, cooperativity and Hebb's cell assemblies: a personal history". Philosophical transactions of the Royal Society of London. Series B, Biological sciences 358 (1432): 629–34. doi:10.1098/rstb.2002.1231. PMC 1693161. PMID 12740107. http://journals.royalsociety.org/openurl.asp?genre=article&issn=0962-8436&volume=358&issue=1432&spage=629.
- ^ Abraham WC (April 2003). "How long will long-term potentiation last?". Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences 358 (1432): 735–44. doi:10.1098/rstb.2002.1222. PMC 1693170. PMID 12740120. http://rstb.royalsocietypublishing.org/cgi/pmidlookup?view=long&pmid=12740120.
- ^ a b c d e f g h i Sweatt J (1999). "Toward a molecular explanation for long-term potentiation". Learn Mem 6 (5): 399–416. doi:10.1101/lm.6.5.399. PMID 10541462. http://www.learnmem.org/cgi/content/full/6/5/399.
- ^ a b c d e f g h i j Lynch M (2004). "Long-term potentiation and memory". Physiol Rev 84 (1): 87–136. doi:10.1152/physrev.00014.2003. PMID 14715912. http://physrev.physiology.org/cgi/content/full/84/1/87.
- ^ Huang Y, Kandel E (1994). "Recruitment of long-lasting and protein kinase A-dependent long-term potentiation in the CA1 region of hippocampus requires repeated tetanization". Learn Mem 1 (1): 74–82. PMID 10467587.
- ^ Agranoff, Bernard W.; Siegel, George J. (1999). Basic neurochemistry: molecular, cellular, and medical aspects. Philadelphia: Lippincott-Raven. pp. 326. ISBN 0-397-51820-X. http://www.ncbi.nlm.nih.gov/books/bv.fcgi?rid=bnchm.section.1131.
- ^ Malinow R (2003). "AMPA receptor trafficking and long-term potentiation". Philos Trans R Soc Lond B Biol Sci 358 (1432): 707–14. doi:10.1098/rstb.2002.1233. PMC 1693162. PMID 12740116. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1693162.
- ^ a b Emptage N, Reid C, Fine A, Bliss T (2003). "Optical quantal analysis reveals a presynaptic component of LTP at hippocampal Schaffer-associational synapses". Neuron 38 (5): 797–804. doi:10.1016/S0896-6273(03)00325-8. PMID 12797963. http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B6WSS-4C5XN9K-G&_coverDate=06%2F05%2F2003&_alid=523934887&_rdoc=1&_fmt=&_orig=search&_qd=1&_cdi=7054&_sort=d&view=c&_acct=C000050221&_version=1&_urlVersion=0&_userid=10&md5=15f6a55d9b53d2265077f3a74a8a495a.
- ^ Frey U, Frey S, Schollmeier F, Krug M (1 January 1996). "Influence of actinomycin D, a RNA synthesis inhibitor, on long-term potentiation in rat hippocampal neurons in vivo and in vitro". J Physiol. 490 (Pt 3) (Pt 3): 703–11. PMC 1158708. PMID 8683469. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1158708.
- ^ Frey U, Krug M, Reymann K, Matthies H (1988). "Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro". Brain Res 452 (1-2): 57–65. doi:10.1016/0006-8993(88)90008-X. PMID 3401749.
- ^ a b c d e f Kelleher R, Govindarajan A, Tonegawa S (2004). "Translational regulatory mechanisms in persistent forms of synaptic plasticity". Neuron 44 (1): 59–73. doi:10.1016/j.neuron.2004.09.013. PMID 15450160.
- ^ Kovács KA, Steullet P, Steinmann M, Do KQ, Magistretti PJ, Halfon O, Cardinaux JR (2007). "TORC1 is a calcium- and cAMP-sensitive coincidence detector involved in hippocampal long-term synaptic plasticity.". PNAS 104 (11): 4700–5. doi:10.1073/pnas.0607524104. PMC 1838663. PMID 17360587. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1838663.
- ^ a b c d e Serrano P, Yao Y, Sacktor T (2005). "Persistent phosphorylation by protein kinase Mzeta maintains late-phase long-term potentiation". J Neurosci 25 (8): 1979–84. doi:10.1523/JNEUROSCI.5132-04.2005. PMID 15728837.
- ^ a b c Pastalkova E, Serrano P, Pinkhasova D, Wallace E, Fenton A, Sacktor T (2006). "Storage of spatial information by the maintenance mechanism of LTP". Science 313 (5790): 1141–4. doi:10.1126/science.1128657. PMID 16931766.
- ^ Kang H, Schuman E (1996). "A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity". Science 273 (5280): 1402–6. doi:10.1126/science.273.5280.1402. PMID 8703078.
- ^ Steward O, Worley P (2001). "A cellular mechanism for targeting newly synthesized mRNAs to synaptic sites on dendrites". Proc Natl Acad Sci USA 98 (13): 7062–8. doi:10.1073/pnas.131146398. PMC 34623. PMID 11416188. http://www.pnas.org/cgi/content/full/98/13/7062.
- ^ Pavlidis P, Montgomery J, Madison D (2000). "Presynaptic protein kinase activity supports long-term potentiation at synapses between individual hippocampal neurons". J Neurosci 20 (12): 4497–505. PMID 10844019.
- ^ Zakharenko S, Patterson S, Dragatsis I, Zeitlin S, Siegelbaum S, Kandel E, Morozov A (2003). "Presynaptic BDNF required for a presynaptic but not postsynaptic component of LTP at hippocampal CA1-CA3 synapses". Neuron 39 (6): 975–90. doi:10.1016/S0896-6273(03)00543-9. PMID 12971897.
- ^ Frey U, Morris R (1997). "Synaptic tagging and long-term potentiation". Nature 385 (6616): 533–6. doi:10.1038/385533a0. PMID 9020359.
- ^ Martin K, Casadio A, Zhu H, Yaping E, Rose J, Chen M, Bailey C, Kandel E (1997). "Synapse-specific, long-term facilitation of aplysia sensory to motor synapses: a function for local protein synthesis in memory storage". Cell 91 (7): 927–38. doi:10.1016/S0092-8674(00)80484-5. PMID 9428516.
- ^ Casadio A, Martin K, Giustetto M, Zhu H, Chen M, Bartsch D, Bailey C, Kandel E (1999). "A transient, neuron-wide form of CREB-mediated long-term facilitation can be stabilized at specific synapses by local protein synthesis". Cell 99 (2): 221–37. doi:10.1016/S0092-8674(00)81653-0. PMID 10535740.
- ^ Segal M, Murphy D (1999). "CREB activation mediates plasticity in cultured hippocampal neurons". Neural Plast 6 (3): 1–7. doi:10.1155/NP.1998.1. PMC 2565317. PMID 9920677. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2565317.
- ^ Straube T, Frey J (2003). "Involvement of beta-adrenergic receptors in protein synthesis-dependent late long-term potentiation (LTP) in the dentate gyrus of freely moving rats: the critical role of the LTP induction strength". Neuroscience 119 (2): 473–9. doi:10.1016/S0306-4522(03)00151-9. PMID 12770561.
- ^ Lu Y, Kandel E, Hawkins R (1999). "Nitric oxide signaling contributes to late-phase LTP and CREB phosphorylation in the hippocampus". J Neurosci 19 (23): 10250–61. PMID 10575022.
- ^ Frey U, Matthies H, Reymann K, Matthies H (1991). "The effect of dopaminergic D1 receptor blockade during tetanization on the expression of long-term potentiation in the rat CA1 region in vitro". Neurosci Lett 129 (1): 111–4. doi:10.1016/0304-3940(91)90732-9. PMID 1833673.
- ^ Otmakhova N, Lisman J (1996). "D1/D5 dopamine receptor activation increases the magnitude of early long-term potentiation at CA1 hippocampal synapses". J Neurosci 16 (23): 7478–86. PMID 8922403.
- ^ Morris R, Anderson E, Lynch G, Baudry M (1986). "Selective impairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, AP5". Nature 319 (6056): 774–6. doi:10.1038/319774a0. PMID 2869411.
- ^ McHugh T, Blum K, Tsien J, Tonegawa S, Wilson M (1996). "Impaired hippocampal representation of space in CA1-specific NMDAR1 knockout mice". Cell 87 (7): 1339–49. doi:10.1016/S0092-8674(00)81828-0. PMID 8980239.
- ^ Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ (1999). "Genetic enhancement of learning and memory in mice". Nature 401 (6748): 63–69. doi:10.1038/43432. PMID 10485705. http://www.nature.com/nature/journal/v401/n6748/abs/401063a0.html.
- ^ Tang Y, Wang H, Feng R, Kyin M, Tsien J (2001). "Differential effects of enrichment on learning and memory function in NR2B transgenic mice". Neuropharmacology 41 (6): 779–90. doi:10.1016/S0028-3908(01)00122-8. PMID 11640933.
- ^ Whitlock J, Heynen A, Shuler M, Bear M (2006). "Learning induces long-term potentiation in the hippocampus". Science 313 (5790): 1093–7. doi:10.1126/science.1128134. PMID 16931756.
- ^ Bliss T, Collingridge G, Laroche S (2006). "Neuroscience. ZAP and ZIP, a story to forget". Science 313 (5790): 1058–9. doi:10.1126/science.1132538. PMID 16931746.
- ^ Cooke SF, Bliss TV (July 2006). "Plasticity in the human central nervous system". Brain: A Journal of Neurology 129 (Pt 7): 1659–73. doi:10.1093/brain/awl082. PMID 16672292. http://brain.oxfordjournals.org/cgi/pmidlookup?view=long&pmid=16672292.
- ^ a b Rowan MJ, Klyubin I, Cullen WK, Anwyl R (April 2003). "Synaptic plasticity in animal models of early Alzheimer's disease". Philosophical transactions of the Royal Society of London. Series B, Biological sciences 358 (1432): 821–8. doi:10.1098/rstb.2002.1240. PMC 1693153. PMID 12740129. http://journals.royalsociety.org/openurl.asp?genre=article&issn=0962-8436&volume=358&issue=1432&spage=821.
- ^ Crary JF, Shao CY, Mirra SS, Hernandez AI, Sacktor TC (April 2006). "Atypical protein kinase C in neurodegenerative disease I: PKMzeta aggregates with limbic neurofibrillary tangles and AMPA receptors in Alzheimer disease". Journal of neuropathology and experimental neurology 65 (4): 319–26. doi:10.1097/01.jnen.0000218442.07664.04. PMID 16691113. http://meta.wkhealth.com/pt/pt-core/template-journal/lwwgateway/media/landingpage.htm?an=00005072-200604000-00002.
- ^ a b Kauer JA, Malenka RC (November 2007). "Synaptic plasticity and addiction". Nature reviews. Neuroscience 8 (11): 844–58. doi:10.1038/nrn2234. PMID 17948030.
- ^ Wolf ME (August 2003). "LTP may trigger addiction". Molecular interventions 3 (5): 248–52. doi:10.1124/mi.3.5.248. PMID 14993438. http://molinterv.aspetjournals.org/cgi/pmidlookup?view=long&pmid=14993438.
Further reading
- Bliss, T; Collingridge, G; Morris, R (2004). Long-term potentiation: enhancing neuroscience for 30 years. Oxford: Oxford University Press. ISBN 0-19-853030-7.
External links
- Researchers provide first evidence for learning mechanism, a PhysOrg.com report on 2006 study by Bear and colleagues.
- Short video documentary about the Doogie mice. (RealPlayer format)
- "Smart Mouse", a Quantum ABC TV episode about the Doogie mice.
- MeSH Long-Term+Potentiation
Nervous system physiology: neurophysiology / clinical neurophysiology Primarily CNS Primarily PNS Both Bereitschaftspotential · P300 · Auditory evoked potential · Somatosensory evoked potentials · Somatosensory evoked potentials · Visual evoked potentialOther short termLong termAxoplasmic transport · Neuroregeneration/Nerve regeneration · Neuroplasticity/Synaptic plasticity (Long-term potentiation, Long-term depression)OtherCategories:- Neurophysiology
- Memory
- Behavioral neuroscience



Wikimedia Foundation. 2010.