- Epilepsy
-
"Epileptic" redirects here. For the graphic novel, see Epileptic (graphic novel)."Epilepsia" redirects here. For the journal, see Epilepsia (journal).
Epilepsy Classification and external resources 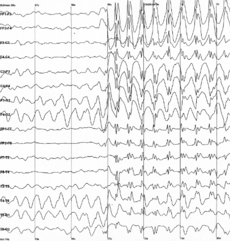
Generalized 3 Hz spike and wave discharges in EEGICD-10 G40-G41 ICD-9 345 DiseasesDB 4366 MedlinePlus 000694 eMedicine neuro/415 MeSH D004827 Epilepsy (from the Ancient Greek ἐπιληψία (epilēpsía) — "seizure") is a common chronic neurological disorder characterized by seizures.[1][2] These seizures are transient signs and/or symptoms of abnormal, excessive or hypersynchronous neuronal activity in the brain.[3] About 50 million people worldwide have epilepsy, and nearly two out of every three new cases are discovered in developing countries.[4] Epilepsy becomes more common as people age.[5][6] Onset of new cases occur most frequently in infants and the elderly.[7] As a consequence of brain surgery, epileptic seizures may occur in recovering patients.
Epilepsy is usually controlled, but not cured, with medication. However, over 30% of people with epilepsy do not have seizure control even with the best available medications. Surgery may be considered in difficult cases.[8][9] Not all epilepsy syndromes are lifelong – some forms are confined to particular stages of childhood. Epilepsy should not be understood as a single disorder, but rather as syndromic with vastly divergent symptoms, all involving episodic abnormal electrical activity in the brain and numerous seizures.
Contents
Classification
Main article: Seizure typesEpilepsies are classified in five ways:
- By their first cause (or etiology).
- By the observable manifestations of the seizures, known as semiology.
- By the location in the brain where the seizures originate.
- As a part of discrete, identifiable medical syndromes.
- By the event that triggers the seizures, such as reading or music
In 1981, the International League Against Epilepsy (ILAE) proposed a classification scheme for individual seizures that remains in common use.[10] This classification is based on observation (clinical and EEG) rather than the underlying pathophysiology or anatomy and is outlined later on in this article. In 1989, the ILAE proposed a classification scheme for epilepsies and epileptic syndromes.[11] This can be broadly described as a two-axis scheme having the cause on one axis and the extent of localization within the brain on the other. Since 1997, the ILAE have been working on a new scheme that has five axes: ictal phenomenon, (pertaining to an epileptic seizure), seizure type, syndrome, etiology, impairment.[12]
Seizure types
Seizure types are organized firstly according to whether the source of the seizure within the brain is localized (partial or focal onset seizures) or distributed (generalized seizures). Partial seizures are further divided on the extent to which consciousness is affected. If it is unaffected, then it is a simple partial seizure; otherwise it is a complex partial (psychomotor) seizure. A partial seizure may spread within the brain - a process known as secondary generalization. Generalized seizures are divided according to the effect on the body but all involve loss of consciousness. These include absence (petit mal), myoclonic, clonic, tonic, tonic-clonic (grand mal), and atonic seizures.[13]
Children may exhibit behaviors that are easily mistaken for epileptic seizures but are not caused by epilepsy. These include:
- Inattentive staring
- Benign shudders (among children younger than age 2, usually when they are tired or excited)
- Self-gratification behaviors (nodding, rocking, head banging)
- Conversion disorder (flailing and jerking of the head, often in response to severe personal stress such as physical abuse)
Conversion disorder can be distinguished from epilepsy because the episodes never occur during sleep and do not involve incontinence or self-injury.[14]
Epilepsy syndromes
Just as there are many types of seizures, there are many types of epilepsy syndromes. Epilepsy classification includes more information about the person and the episodes than seizure type alone, such as clinical features (e.g., behavior during the seizure) and expected causes.[15]
There are four main groups of epileptic syndrome which can be further divided into: benign Rolandic epilepsy, frontal lobe epilepsy, infantile spasms, juvenile myoclonic epilepsy, juvenile absence epilepsy, childhood absence epilepsy (pyknolepsy), hot water epilepsy, Lennox-Gastaut syndrome, Landau-Kleffner syndrome, mitochondrial disorders, progressive myoclonic epilepsy, reflex epilepsy, Rasmussen's syndrome, temporal lobe epilepsy, limbic epilepsy, status epilepticus, abdominal epilepsy, massive bilateral myoclonus, catamenial epilepsy, Jacksonian seizure disorder, Lafora disease, photosensitive epilepsy, etc.
Each type of epilepsy presents with its own unique combination of seizure type, typical age of onset, EEG findings, treatment, and prognosis. The most widespread classification of the epilepsies [11] divides epilepsy syndromes by location or distribution of seizures (as revealed by the appearance of the seizures and by EEG) and by cause. Syndromes are divided into localization-related epilepsies, generalized epilepsies, or epilepsies of unknown localization.
Localization-related epilepsies, sometimes termed partial or focal epilepsies, arise from an epileptic focus, a small portion of the brain that serves as the irritant driving the epileptic response. Generalized epilepsies, in contrast, arise from many independent foci (multifocal epilepsies) or from epileptic circuits that involve the whole brain. Epilepsies of unknown localization remain unclear as to whether they arise from a portion of the brain or from more widespread circuits.
Epilepsy syndromes are further divided by presumptive cause: idiopathic, symptomatic, and cryptogenic. In general, idiopathic epilepsies are thought to arise from genetic abnormalities that lead to alteration of basic neuronal regulation.[16] Symptomatic epilepsies arise from the effects of an epileptic lesion, whether that lesion is focal, such as a tumor, or a defect in metabolism causing widespread injury to the brain. Cryptogenic epilepsies involve a presumptive lesion that is otherwise difficult or impossible to uncover during evaluation.
The genetic component to epilepsy is receiving particular interest from the scientific community. Conditions such as ring chromosome 20 syndrome (r(20)) are gaining acknowledgment, and although only 60 cases have been reported in the literature since 1976, "more widespread cytogenetic chromosomal karyotyping in nonetiological cases of epilepsy" is likely.[17]
Some epileptic syndromes are difficult to fit within this classification scheme and fall in the unknown localization/etiology category. People with seizures that occur only after specific precipitants ("provoked seizures"), have "epilepsies" that fall into this category. Febrile convulsions are an example of seizures bound to a particular precipitant. Landau-Kleffner syndrome is another epilepsy that, because of its variety of EEG distributions, falls uneasily in clear categories. What can be even more confusing is that certain syndromes, such as West syndrome, featuring seizures such as infantile spasms, can be classified as idiopathic, syndromic, or cryptogenic depending on cause and can arise from both focal or generalized epileptic lesions.
Below are some common seizure syndromes:
- Autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) is an idiopathic localization-related epilepsy that is an inherited epileptic disorder that causes seizures during sleep. Onset is usually in childhood. These seizures arise from the frontal lobes and consist of complex motor movements, such as hand clenching, arm raising/lowering, and knee bending. Vocalizations such as shouting, moaning, or crying are also common. ADNFLE is often misdiagnosed as nightmares. ADNFLE has a genetic basis.[18] These genes encode various nicotinic acetylcholine receptors.
- Benign centrotemporal lobe epilepsy of childhood or benign Rolandic epilepsy is an idiopathic localization-related epilepsy that occurs in children between the ages of 3 and 13 years, with peak onset in prepubertal late childhood. Apart from their seizure disorder, these patients are otherwise normal. This syndrome features simple partial seizures that involve facial muscles and frequently cause drooling. Although most episodes are brief, seizures sometimes spread and generalize. Seizures are typically nocturnal and confined to sleep. The EEG may demonstrate spike discharges that occur over the centrotemporal scalp over the central sulcus of the brain (the Rolandic sulcus) that are predisposed to occur during drowsiness or light sleep. Seizures cease near puberty.[19] Seizures may require anticonvulsant treatment, but sometimes are infrequent enough to allow physicians to defer treatment.
- Benign occipital epilepsy of childhood (BOEC) is an idiopathic localization-related epilepsy and consists of an evolving group of syndromes. Most authorities include two subtypes, an early subtype with onset between three and five years, and a late onset between seven and 10 years. Seizures in BOEC usually feature visual symptoms such as scotoma or fortifications (brightly colored spots or lines) or amaurosis (blindness or impairment of vision). Convulsions involving one half the body, hemiconvulsions, or forced eye deviation or head turning are common. Younger patients typically experience symptoms similar to migraine with nausea and headache, and older patients typically complain of more visual symptoms. The EEG in BOEC shows spikes recorded from the occipital (back of head) regions. The EEG and genetic pattern suggest an autosomal dominant transmission as described by Ruben Kuzniecky, et al.[20] Lately, a group of epilepsies termed Panayiotopoulos syndrome[21] that share some clinical features of BOEC but have a wider variety of EEG findings are classified by some as BOEC.
- Catamenial epilepsy (CE) is when seizures cluster around certain phases of a woman's menstrual cycle.
- Childhood absence epilepsy (CAE) is an idiopathic generalized epilepsy that affects children between the ages of 4 and 12 years of age, although peak onset is around five to six years old. These patients have recurrent absence seizures, brief episodes of unresponsive staring, sometimes with minor motor features such as eye blinking or subtle chewing. The EEG finding in CAE is generalized 3 Hz spike and wave discharges. Some go on to develop generalized tonic-clonic seizures. This condition carries a good prognosis because children do not usually show cognitive decline or neurological deficits, and the seizures in the majority cease spontaneously with onging maturation.
- Dravet's syndrome, previously known as severe myoclonic epilepsy of infancy (SMEI), is a neurodevelopmental disorder beginning in infancy and characterized by severe epilepsy that does not respond well to treatment. This syndrome was described by Charlotte Dravet, French psychiatrist and epileptologist (born July 14, 1936). Dravet described this syndrome while working at the Centre Saint Paul at the University of Marseille. At Centre Saint Paul, one of her supervisors was Henri Gastaut, who described the Lennox-Gastaut syndrome. She described this condition in 1978 [22] Estimates of the prevalence of this rare disorder have ranged from 1:20,000 to 1:40,000 births, though the incidence may be found to be greater as the syndrome becomes better recognized and new genetic evidence is discovered. It is thought to occur with similar frequency in both genders, and knows no geographic or ethnic boundaries.
- The course of Dravet syndrome is highly variable from person to person. Seizures begin during the first year of life and development is normal prior to their onset. In most cases, the first seizures occur with fever and are generalized tonic-clonic (grand mal) or unilateral (one-sided) convulsions. These seizures are often prolonged, and may lead to status epilepticus, a medical emergency. In time, seizures increase in frequency and begin to occur without fever. Additional seizure types appear, most often these are myoclonic, atypical absence, and complex-partial seizures.
- Additional features that are seen in significant numbers of patients with Dravet syndrome may include sensory integration disorders and other autism spectrum characteristics, orthopedic or movement disorders, frequent or chronic upper respiratory and ear infections, sleep disturbance, dysautonomia, and problems with growth and nutrition.[23]
- Frontal lobe epilepsy, usually a symptomatic or cryptogenic localization-related epilepsy, arises from lesions causing seizures that occur in the frontal lobes of the brain. These epilepsies can be difficult to diagnose because the symptoms of seizures can easily be confused with nonepileptic spells and, because of limitations of the EEG, be difficult to "see" with standard scalp EEG.
- Juvenile absence epilepsy is an idiopathic generalized epilepsy with later onset than CAE, typically in prepubertal adolescence, with the most frequent seizure type being absence seizures. Generalized tonic-clonic seizures can occur. Often, 3 Hz spike-wave or multiple spike discharges can be seen on EEG. The prognosis is mixed, with some patients going on to a syndrome that is poorly distinguishable from JME.
- Juvenile myoclonic epilepsy (JME) is an idiopathic generalized epilepsy that occurs in patients aged 8 to 20 years. Patients have normal cognition and are otherwise neurologically intact. The most common seizures are myoclonic jerks, although generalized tonic-clonic seizures and absence seizures may occur as well. Myoclonic jerks usually cluster in the early morning after awakening. The EEG reveals generalized 4–6 Hz spike wave discharges or multiple spike discharges. Interestingly, these patients are often first diagnosed when they have their first generalized tonic-clonic seizure later in life, when they experience sleep deprivation (e.g., freshman year in college after staying up late to study for exams). Alcohol withdrawal can also be a major contributing factor in breakthrough seizures, as well. The risk of the tendency to have seizures is lifelong; however, the majority have well-controlled seizures with anticonvulsant medication and avoidance of seizure precipitants.
- Lennox-Gastaut syndrome (LGS) is a generalized epilepsy that consists of a triad of developmental delay or childhood dementia, mixed generalized seizures, and EEG demonstrating a pattern of approximately 2 Hz "slow" spike-waves. Onset occurs between two and 18 years. As in West syndrome, LGS result from idiopathic, symptomatic, or cryptogenic causes, and many patients first have West syndrome. Authorities emphasize different seizure types as important in LGS, but most have astatic seizures (drop attacks), tonic seizures, tonic-clonic seizures, atypical absence seizures, and sometimes, complex partial seizures. Anticonvulsants are usually only partially successful in treatment.
- Ohtahara syndrome is a rare, but severe epilepsy syndrome usually starting in the first few days or weeks of life. The seizures are often in the form of stiffening spasms but other seizures including unilateral ones may be seen. The electroencephalogram (EEG) is characteristic. The prognosis is poor with about half of the infants dying in the first year of life; most if not all surviving infants are severely intellectually disabled and many have cerebral palsy. There is no effective treatment. A number of children have underlying structural brain abnormalities.[24]
- Primary reading epilepsy is a reflex epilepsy classified as an idiopathic localization-related epilepsy. Reading in susceptible individuals triggers characteristic seizures.[25]
- Progressive myoclonic epilepsies define a group of symptomatic generalized epilepsies characterized by progressive dementia and myoclonic seizures. Tonic-clonic seizures may occur as well. Diseases usually classified in this group are Unverricht-Lundborg disease, myoclonus epilepsy with ragged red fibers (MERRF syndrome), Lafora disease, neuronal ceroid lipofucinosis, and sialdosis.
- Rasmussen's encephalitis is a symptomatic localization-related epilepsy that is a progressive, inflammatory lesion affecting children with onset before the age of 10. Seizures start as separate simple partial or complex partial seizures and may progress to epilepsia partialis continua (simple partial status epilepticus). Neuroimaging shows inflammatory encephalitis on one side of the brain that may spread if not treated. Dementia and hemiparesis are other problems. The cause is hypothesized to involve an immulogical attack against glutamate receptors, a common neurotransmitter in the brain.[26]
- Symptomatic localization-related epilepsies are divided by the location in the brain of the epileptic lesion, since the symptoms of the seizures are more closely tied to the brain location rather than the cause of the lesion. Tumors, atriovenous malformations, cavernous malformations, trauma, and cerebral infarcts can all be causes of epileptic foci in different brain regions.
- Temporal lobe epilepsy (TLE), a symptomatic localization-related epilepsy, is the most common epilepsy of adults who experience seizures poorly controlled with anticonvulsant medications. In most cases, the epileptogenic region is found in the midline (mesial) temporal structures (e.g., the hippocampus, amygdala, and parahippocampal gyrus). Seizures begin in late childhood and adolescence. Most of these patients have complex partial seizures sometimes preceded by an aura, and some TLE patients also suffer from secondary generalized tonic-clonic seizures. If the patient does not respond sufficiently to medical treatment, epilepsy surgery may be considered.
- Tuberous Sclerosis (TSC) is a genetic disorder that causes tumors to form in many different organs, primarily in the brain, eyes, heart, kidney, skin and lungs. Several types of brain lesions can occur in individuals with TSC and 60% - 90% of people with TSC develop epilepsy.
- West syndrome is a triad of developmental delay, seizures termed infantile spasms, and EEG demonstrating a pattern termed hypsarrhythmia. Onset occurs between three months and two years, with peak onset between eight and 9 months. West syndrome may arise from idiopathic, symptomatic, or cryptogenic causes. The most common cause is tuberous sclerosis. The prognosis varies with the underlying cause. In general, most surviving patients remain with significant cognitive impairment and continuing seizures and may evolve to another eponymic syndrome, Lennox-Gastaut syndrome.
Causes
The diagnosis of epilepsy usually requires that the seizures occur spontaneously. Nevertheless, certain epilepsy syndromes require particular precipitants or triggers for seizures to occur. These are termed reflex epilepsy. For example, patients with primary reading epilepsy have seizures triggered by reading. Photosensitive epilepsy can be limited to seizures triggered by flashing lights. Other precipitants can trigger an epileptic seizure in patients who otherwise would be susceptible to spontaneous seizures. For example, children with childhood absence epilepsy may be susceptible to hyperventilation. In fact, flashing lights and hyperventilation are activating procedures used in clinical EEG to help trigger seizures to aid diagnosis. Finally, other precipitants can facilitate, rather than obligately trigger, seizures in susceptible individuals. Emotional stress, sleep deprivation, sleep itself, heat stress, alcohol and febrile illness are examples of precipitants cited by patients with epilepsy. Notably, the influence of various precipitants varies with the epilepsy syndrome.[27] Likewise, the menstrual cycle in women with epilepsy can influence patterns of seizure recurrence. Catamenial epilepsy is the term denoting seizures linked to the menstrual cycle.[28]
There are different causes of epilepsy that are common in certain age groups.
- During the neonatal period and early infancy the most common causes include hypoxic-ischemic encephalopathy, CNS infections, trauma, congenital CNS abnormalities, and metabolic disorders.
- During late infancy and early childhood, febrile seizures are fairly common. These may be caused by many different things, some thought to be things such as CNS infections and trauma.
- During childhood, well-defined epilepsy syndromes are generally seen.
- During adolescence and adulthood, the causes are more likely to be secondary to any CNS lesion. Further, idiopathic epilepsy is less common. Other causes associated with these age groups are stress, trauma, CNS infections, brain tumors, illicit drug use and alcohol withdrawal.
- In older adults, cerebrovascular disease is a very common cause. Other causes are CNS tumors, head trauma, and other degenerative diseases that are common in the older age group, such as dementia.[29]
Pathophysiology
Mutations in several genes have been linked to some types of epilepsy. Several genes that code for protein subunits of voltage-gated and ligand-gated ion channels have been associated with forms of generalized epilepsy and infantile seizure syndromes.[30]
One speculated mechanism for some forms of inherited epilepsy are mutations of the genes that code for sodium channel proteins; these defective sodium channels stay open for too long, thus making the neuron hyper-excitable. Glutamate, an excitatory neurotransmitter, may, therefore, be released from these neurons in large amounts, which — by binding with nearby glutamatergic neurons — triggers excessive calcium (Ca2+) release in these post-synaptic cells. Such excessive calcium release can be neurotoxic to the affected cell. The hippocampus, which contains a large volume of just such glutamatergic neurons (and NMDA receptors, which are permeable to Ca2+ entry after binding of both glutamate and glycine), is especially vulnerable to epileptic seizure, subsequent spread of excitation, and possible neuronal death. Another possible mechanism involves mutations leading to ineffective GABA (the brain's most common inhibitory neurotransmitter) action. Epilepsy-related mutations in some non-ion channel genes have also been identified.
Much like the channelopathies in voltage-gated ion channels, several ligand-gated ion channels have been linked to some types of frontal and generalized epilepsies.
Epileptogenesis is the process by which a normal brain develops epilepsy after trauma, such as a lesion on the brain. One interesting finding in animals is that repeated low-level electrical stimulation to some brain sites can lead to permanent increases in seizure susceptibility: in other words, a permanent decrease in seizure "threshold." This phenomenon, known as kindling (by analogy with the use of burning twigs to start a larger fire) was discovered by Dr. Graham Goddard in 1967. It is important to note that these "kindled" animals do not experience spontaneous seizures. Chemical stimulation can also induce seizures; repeated exposures to some pesticides have been shown to induce seizures in both humans and animals. One mechanism proposed for this is called excitotoxicity. The roles of kindling and excitotoxicity, if any, in human epilepsy are currently hotly debated.
Other causes of epilepsy are brain lesions, where there is scar tissue or another abnormal mass of tissue in an area of the brain.
The complexity of understanding what seizures are have led to considerable efforts to use computational models of epilepsy to both interpret experimental and clinical data, as well as guide strategies for therapy.
Physical, emotional, and social functioning of youth are interfered specifically if seizures are uncontrolled.[31] Some other noted consequences on repeated seizures are neuronal loss, gliosis, parenhymal microhemorrhages, excess of starch bodies, leptomeningeal thickening, subpial gliosis, perivascular gliosis and periavascular atrophy.[32]
Management
Epilepsy is usually treated with medication prescribed by a physician; primary caregivers, neurologists, and neurosurgeons all frequently care for people with epilepsy. However, it has been stressed that accurate differentiation between generalized and partial seizures is especially important in determining the appropriate treatment.[33] In some cases the implantation of a stimulator of the vagus nerve, or a special diet can be helpful. Neurosurgical operations for epilepsy can be palliative, reducing the frequency or severity of seizures; or, in some patients, an operation can be curative.[citation needed]
The proper initial response to a generalized tonic-clonic epileptic seizure is to roll the person on the side (recovery position) to prevent ingestion of fluids into the lungs, which can result in choking and death. Should the person regurgitate, this should be allowed to drip out the side of the person's mouth. The person should be prevented from self-injury by moving them away from sharp edges, and placing something soft beneath the head. If a seizure lasts longer than 5 minutes, or if more than one seizure occurs without regaining consciousness emergency medical services should be contacted.[citation needed]
Medications
Main article: AnticonvulsantThe mainstay of treatment of epilepsy is anticonvulsant medications. Often, anticonvulsant medication treatment will be lifelong and can have major effects on quality of life. The choice among anticonvulsants and their effectiveness differs by epilepsy syndrome. Mechanisms, effectiveness for particular epilepsy syndromes, and side-effects differ among the individual anticonvulsant medications. Some general findings about the use of anticonvulsants are outlined below.
Availability - Currently there are 20 medications approved by the Food and Drug Administration for the use of treatment of epileptic seizures in the US: carbamazepine (common US brand name Tegretol), clorazepate (Tranxene), clonazepam (Klonopin), ethosuximide (Zarontin), felbamate (Felbatol), fosphenytoin (Cerebyx), gabapentin (Neurontin), lacosamide (Vimpat), lamotrigine (Lamictal), levetiracetam (Keppra), oxcarbazepine (Trileptal), phenobarbital (Luminal), phenytoin (Dilantin), pregabalin (Lyrica), primidone (Mysoline), tiagabine (Gabitril), topiramate (Topamax), valproate semisodium (Depakote), valproic acid (Depakene), and zonisamide (Zonegran). Most of these appeared after 1990.
Medications commonly available outside the US but still labelled as "investigational" within the US are clobazam (Frisium) and vigabatrin (Sabril). Medications currently under clinical trial under the supervision of the FDA include retigabine, brivaracetam, and seletracetam.
Other drugs are commonly used to abort an active seizure or interrupt a seizure flurry; these include diazepam (Valium, Diastat) and lorazepam (Ativan). Drugs used only in the treatment of refractory status epilepticus include paraldehyde (Paral), midazolam (Versed), and pentobarbital (Nembutal).
Some anticonvulsant medications do not have primary FDA-approved uses in epilepsy but are used in limited trials, remain in rare use in difficult cases, have limited "grandfather" status, are bound to particular severe epilepsies, or are under current investigation. These include acetazolamide (Diamox), progesterone, adrenocorticotropic hormone (ACTH, Acthar), various corticotropic steroid hormones (prednisone), or bromide.
Effectiveness - The definition of "effective" varies. FDA approval usually requires that 50% of the patient treatment group had at least a 50% improvement in the rate of epileptic seizures. About 20% of patients with epilepsy continue to have breakthrough epileptic seizures despite best anticonvulsant treatment.[8][9]
Safety and Side Effects - 88% of patients with epilepsy, in a European survey, reported at least one anticonvulsant related side-effect.[34] Most side effects are mild and "dose-related" and can often be avoided or minimized by the use of the smallest effective amount. Some examples include mood changes, sleepiness, or unsteadiness in gait. Some anticonvulsant medications have "idiosyncratic" side effects that can not be predicted by dose. Some examples include drug rashes, liver toxicity (hepatitis), or aplastic anemia. Safety includes the consideration of teratogenicity (the effects of medications on fetal development) when women with epilepsy become pregnant.
Principles of Anticonvulsant Use and Management - The goal for individual patients is no seizures and minimal side-effects, and the job of the physician is to aid the patient to find the best balance between the two during the prescribing of anticonvulsants. Most patients can achieve this balance best with monotherapy, the use of a single anticonvulsant medication. Some patients, however, require polypharmacy, the use of two or more anticonvulsants.
Serum levels of AEDs can be checked to determine medication compliance, to assess the effects of new drug-drug interactions upon previous stable medication levels, or to help establish if particular symptoms such as instability or sleepiness can be considered a drug side effect or are due to different causes. Children or impaired adults who may not be able to communicate side-effects may benefit from routine screening of drug levels. Beyond baseline screening, however, trials of recurrent, routine blood or urine monitoring show no proven benefits and may lead to unnecessary medication adjustments in most older children and adults using routine anticonvulsants.[35][36]
If a person's epilepsy cannot be brought under control after adequate trials of two or three (experts vary here) different drugs, that person's epilepsy is generally said to be medically refractory. A study of patients with previously untreated epilepsy demonstrated that 47% achieved control of seizures with the use of their first single drug. 14% became seizure free during treatment with a second or third drug. An additional 3% became seizure-free with the use of two drugs simultaneously.[37] Other treatments, in addition to or instead of, anticonvulsant medications may be considered by those people with continuing seizures.
Surgery
Epilepsy surgery is an option for people with focal seizures that remain resistant to treatment.[38] The goal for these procedures is total control of epileptic seizures,[39] although anticonvulsant medications may still be required.[40]
The evaluation for epilepsy surgery is designed to locate the "epileptic focus" (the location of the epileptic abnormality) and to determine if resective surgery will affect normal brain function. Physicians will also confirm the diagnosis of epilepsy to make sure that spells arise from epilepsy (as opposed to non-epileptic seizures). The evaluation typically includes neurological examination, routine EEG, Long-term video-EEG monitoring, neuropsychological evaluation, and neuroimaging such as MRI, Single photon emission computed tomography (SPECT), positron emission tomography (PET). Some epilepsy centers use intracarotid sodium amobarbital test (Wada test), functional MRI or Magnetoencephalography (MEG) as supplementary tests.
Certain lesions require Long-term video-EEG monitoring with the use of intracranial electrodes if noninvasive testing was inadequate to identify the epileptic focus or distinguish the surgical target from normal brain tissue and function. Brain mapping by the technique of cortical electrical stimulation or Electrocorticography are other procedures used in the process of invasive testing in some patients.
The most common surgeries are the resection of lesions like tumors or arteriovenous malformations, which, in the process of treating the underlying lesion, often result in control of epileptic seizures caused by these lesions.
Other lesions are more subtle and feature epilepsy as the main or sole symptom. The most common form of intractable epilepsy in these disorders in adults is temporal lobe epilepsy with hippocampal sclerosis, and the most common type of epilepsy surgery is the anterior temporal lobectomy, or the removal of the front portion of the temporal lobe including the amygdala and hippocampus. Some neurosurgeons recommend selective amygdalahippocampectomy because of possible benefits in postoperative memory or language function. Surgery for temporal lobe epilepsy is effective, durable, and results in decreased health care costs.[41][42] Despite the efficacy of epilepsy surgery, some patients decide not to undergo surgery owing to fear or the uncertainty of having a brain operation.
Palliative surgery for epilepsy is intended to reduce the frequency or severity of seizures. Examples are callosotomy or commissurotomy to prevent seizures from generalizing (spreading to involve the entire brain), which results in a loss of consciousness. This procedure can therefore prevent injury due to the person falling to the ground after losing consciousness. It is performed only when the seizures cannot be controlled by other means. Multiple subpial transection can also be used to decrease the spread of seizures across the cortex especially when the epileptic focus is located near important functional areas of the cortex. Resective surgery can be considered palliative if it is undertaken with the expectation that it will reduce but not eliminate seizures.
Hemispherectomy involves removal or a functional disconnection of most or all of one half of the cerebrum. It is reserved for people suffering from the most catastrophic epilepsies, such as those due to Rasmussen syndrome. If the surgery is performed on very young patients (2–5 years old), the remaining hemisphere may acquire some rudimentary motor control of the ipsilateral body; in older patients, paralysis results on the side of the body opposite to the part of the brain that was removed. Because of these and other side-effects, it is usually reserved for patients having exhausted other treatment options.
Other
A ketogenic diet (high-fat, low-carbohydrate) was first tested in the 1920s, but became less used with the advent of effective anticonvulsants. In the 1990s specialized diets again gained traction within the medical community.[43] The mechanism of action is unknown. It is used mainly in the treatment of children with severe, medically intractable epilepsies, and the New York Times reported that use is supported by peer-reviewed research that found that the diet reduced seizures among drug-resistant epileptics by >50% in 38% of patients and by >90% in 7% of patients.[44]
While far from a cure, operant-based biofeedback based on conditioning of EEG waves has some experimental support (see Professional practice of behavior analysis). Overall, the support is based on a handful of studies reviewed by Barry Sterman.[45] These studies report a 30% reduction in weekly seizures.
Electrical stimulation [46] methods of anticonvulsant treatment are both currently approved for treatment and investigational uses. A currently approved device is vagus nerve stimulation (VNS). Investigational devices include the responsive neurostimulation system (RNS) and deep brain stimulation (DBS).
- Vagus nerve stimulation (US manufacturer Cyberonics) consists of a computerized electrical device similar in size, shape and implant location to a heart pacemaker that connects to the vagus nerve in the neck. The device stimulates the vagus nerve at preset intervals and intensities of current. Efficacy has been tested in patients with localization-related epilepsies, demonstrating 50% of patients experience a 50% improvement in seizure rate. Case series have demonstrated similar efficacies in certain generalized epilepsies, such as Lennox-Gastaut syndrome. Although success rates are not usually equal to that of epilepsy surgery, it is a reasonable alternative when the patient is reluctant to proceed with any required invasive monitoring, when appropriate presurgical evaluation fails to uncover the location of epileptic foci, or when there are multiple epileptic foci.
- Responsive neurostimulator system (US manufacturer Neuropace) consists of a computerized electrical device implanted in the skull, with electrodes implanted in presumed epileptic foci within the brain. The brain electrodes send EEG signals to the device, which contains seizure-detection software. When certain EEG seizure criteria are met, the device delivers a small electrical charge to other electrodes near the epileptic focus, which disrupt the seizure. The efficacy of the RNS is under current investigation with the goal of FDA approval.
- Deep brain stimulation (US manufacturer Medtronic) consists of a computerized electrical device implanted in the chest in a manner similar to the VNS, but electrical stimulation is delivered to deep brain structures through depth electrodes implanted through the skull. In epilepsy, the electrode target is the anterior nucleus of the thalamus. The efficacy of the DBS in localization-related epilepsies is currently under investigation.
Noninvasive surgery using the gamma knife or other devices used in radiosurgery is currently being investigated as an alternative to traditional open surgery in patients who would otherwise qualify for anterior temporal lobectomy.[47]
Avoidance therapy consists of minimizing or eliminating triggers in patients whose seizures are particularly susceptible to seizure precipitants (see above). For example, sunglasses that counter exposure to particular light wavelengths can improve seizure control in certain photosensitive epilepsies.[48]
Canine warning system is where a seizure response dog, a form of service dog, is trained to summon help or ensure personal safety when a seizure occurs. These are not suitable for everybody, and not all dogs can be so trained. Rarely, a dog may develop the ability to sense a seizure before it occurs.[49] Development of electronic forms of seizure detection systems are currently under investigation.
Seizure prediction-based devices using long-term EEG recordings is presently being evaluated as a new way to stop epileptic seizures before they appear clinically.
Alternative or complementary medicine, including acupuncture,[50] psychological interventions,[51] vitamins[52] and yoga,[53] was evaluated in a number of systematic reviews by the Cochrane Collaboration into treatments for epilepsy, and found there is no reliable evidence to support the use of these as treatments for epilepsy. Exercise or other physical activity[54] [55] have also been proposed as efficacious strategies for preventing or treating epilepsy.
Epidemiology
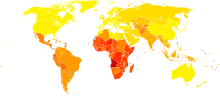 Disability-adjusted life year for epilepsy per 100,000 inhabitants in 2002.
Disability-adjusted life year for epilepsy per 100,000 inhabitants in 2002. no dataless than 5050-72.572.5-9595-117.5117.5-140140-162.5162.5-185185-207.5207.5-230230-252.5252.5-275more than 275
no dataless than 5050-72.572.5-9595-117.5117.5-140140-162.5162.5-185185-207.5207.5-230230-252.5252.5-275more than 275Epilepsy is one of the most common of the serious neurological disorders.[56] About 3% of people will be diagnosed with epilepsy at some time in their lives.[57] Genetic, congenital, and developmental conditions are mostly associated with it among younger patients; tumors are more likely over age 40; head trauma and central nervous system infections may occur at any age. The prevalence of active epilepsy is roughly in the range 5–10 per 1000 people. Up to 5% of people experience non febrile seizures at some point in life; epilepsy's lifetime prevalence is relatively high because most patients either stop having seizures or (less commonly) die of it. Epilepsy's approximate annual incidence rate is 40–70 per 100,000 in industrialized countries and 100–190 per 100,000 in resource-poor countries; socioeconomically deprived people are at higher risk. In industrialized countries the incidence rate decreased in children but increased among the elderly during the three decades prior to 2003, for reasons not fully understood.[58]
Sudden unexpected death in epilepsy
Beyond symptoms of the underlying diseases that can be a part of certain epilepsies, people with epilepsy are at risk for death from four main problems: status epilepticus (most often associated with anticonvulsant noncompliance), suicide associated with depression, trauma from seizures, and sudden unexpected death in epilepsy (SUDEP) [59][60][61] Those at highest risk for epilepsy-related deaths usually have underlying neurological impairment or poorly controlled seizures; those with more benign epilepsy syndromes have little risk for epilepsy-related death.
The NICE National Sentinel Audit of Epilepsy-Related Deaths,[62] led by "Epilepsy Bereaved" drew attention to this important problem. The Audit revealed; "1,000 deaths occur every year in the UK as a result of epilepsy" and most of them are associated with seizures and 42% of deaths were potentially avoidable".[63]
Certain diseases also seem to occur in higher than expected rates in people with epilepsy, and the risk of these "comorbidities" often varies with the epilepsy syndrome. These diseases include depression and anxiety disorders, migraine and other headaches, infertility and low sexual libido. Attention-deficit/hyperactivity disorder (ADHD) affects three to five times more children with epilepsy than children in the general population. [64] ADHD and epilepsy have significant consequences on a child's behavioral, learning, and social development.[65] Epilepsy is prevalent in autism.[66]
History
The word epilepsy is derived from the Ancient Greek ἐπιληψία epilēpsía, which was from ἐπιλαμβάνειν ēpilambánein "to take hold of, to seize", which in turn was combined from ἐπί ēpí "upon" and λαμβάνειν lambánein "to take".[67]
In the past, epilepsy was associated with religious experiences and even demonic possession. In ancient times, epilepsy was known as the "Sacred Disease" (as described in a 5th century BC treatise by Hippocrates[68] ) because people thought that epileptic seizures were a form of attack by demons, or that the visions experienced by persons with epilepsy were sent by the gods. Among animist Hmong families, for example, epilepsy was understood as an attack by an evil spirit, but the affected person could become revered as a shaman through these otherworldly experiences.[69]
A chapter from a Babylonian textbook of medicine, dating from about 2000BC and consisting of 40 tablets, records many of the different seizure types we recognize today, and it emphasizes the supernatural nature of epilepsy,[68] while the Ayurvedic text of Charaka Samhita (about 400BC), describes epilepsy as "apasmara", i.e., "loss of consciousness".[68]
In most cultures, persons with epilepsy have been stigmatized, shunned, or even imprisoned; in the Salpêtrière, the birthplace of modern neurology, Jean-Martin Charcot found people with epilepsy side-by-side with the mentally retarded, those with chronic syphilis, and the criminally insane. In Tanzania to this day, as with other parts of Africa, epilepsy is associated with possession by evil spirits, witchcraft, or poisoning and is believed by many to be contagious.[70] In ancient Rome, epilepsy was known as the Morbus Comitialis ('disease of the assembly hall') and was seen as a curse from the gods.
Stigma continues to this day, in both the public and private spheres, but polls suggest it is generally decreasing with time, at least in the developed world; Hippocrates remarked that epilepsy would cease to be considered divine the day it was understood.[71]
Society and culture
Legal implications
Many jurisdictions forbid certain activities to persons suffering from epilepsy. The most commonly prohibited activities involve operation of vehicles or machinery, or other activities in which continuous vigilance is required. However, there are usually exceptions for those who can prove that they have stabilized their condition. Those few whose seizures do not cause impairment of consciousness, have a lengthy aura preceding impairment of consciousness, or whose seizures only arise from sleep, may be exempt from such restrictions, depending on local laws. There is an ongoing debate in bioethics over who should bear the burden of ensuring that an epilepsy patient does not drive a car or fly an airplane.
Automobiles
Main article: Epilepsy and drivingIn the U.S., people with epilepsy can drive if their seizures are controlled with treatment and they meet the licensing requirements in their state. The amount of time someone needs to be free of seizures varies in different states, but is most likely to be between three months and a year.[72][73] The majority of the 50 states place the burden on patients to report their condition to appropriate licensing authorities so that their privileges can be revoked where appropriate. A minority of states place the burden of reporting on the patient's physician. After reporting is carried out, it is usually the driver's licensing agency that decides to revoke or restrict a driver's license.
In the UK, it is the responsibility of the patients to inform the Driver and Vehicle Licensing Agency (DVLA) if they have epilepsy.[74] The DVLA rules are quite complex,[75] but in summary,[76] those who continue to have seizures or who are within 6 months of medication change may have their licence revoked. A person must be seizure free of an 'awake' seizure for 12 months (or had only 'sleep' seizures for 3 years or more) before they can apply for a license.[77] A doctor who becomes aware that a patient with uncontrolled epilepsy is continuing to drive has, after reminding the patient of their responsibility, a duty to break confidentiality and inform the DVLA. The doctor should advise the patient of the disclosure and the reasons why their failure to notify the agency obliged the doctor to act.
Aircraft
In many countries, persons with any history of epilepsy are generally disqualified for the medical certifications required for all classes of pilot licenses. In the United States, FAA regulations disqualify applicants for medical certification with a history of epilepsy, although the final decision is made by FAA headquarters, and rare exceptions are sometimes made for persons who have had only an isolated seizure or two in childhood and have remained free of seizures in adulthood without medication.[78][79]
In the United Kingdom, a sub-class of pilots license called the National Private Pilot's License has the same medical requirement standards as the DVLA motoring requirements, hence epilepsy sufferers with one year absence free can, with certain exceptions, fly over UK airspace in certain types of aircraft.[80]
Notable cases
Main article: List of people with epilepsyMany notable people, past and present, have carried the diagnosis of epilepsy. In many cases, their epilepsy is a footnote to their accomplishments; for some, it played an integral role in their fame. Historical diagnoses of epilepsy are not always certain; there is controversy about what is considered an acceptable amount of evidence in support of such a diagnosis.
In other animals
Main article: Epilepsy in animalsEpilepsy occurs in a number of other animals including dogs and cats.[81] It is the most common brain disorder in dogs.[81]
See also
- Epilepsy in children
- Epilepsy Phenome/Genome Project
- International Dravet Epilepsy Action League
- ISAS (Ictal-Interictal SPECT Analysis by SPM)
- Issues for epileptics
- On the Sacred Disease
- Paroxysmal depolarizing shift
- Post-traumatic epilepsy
- Psychogenic non-epileptic seizures
- Pyridoxine-dependent epilepsy
- Seizure trigger
- Sudden unexpected death in epilepsy
- The Spirit Catches You and You Fall Down
References
- ^ Commission on Epidemiology and Prognosis, International League Against Epilepsy (1993). "Guidelines for epidemiologic studies on epilepsy. Commission on Epidemiology and Prognosis, International League Against Epilepsy". Epilepsia 34 (4): 592–6. doi:10.1111/j.1528-1157.1993.tb00433.x. PMID 8330566. http://www.blackwell-synergy.com/doi/pdf/10.1111/j.1528-1157.1993.tb00433.x.
- ^ Blume W, Lüders H, Mizrahi E, Tassinari C, van Emde Boas W, Engel J (2001). "Glossary of descriptive terminology for ictal semiology: report of the ILAE task force on classification and terminology". Epilepsia 42 (9): 1212–8. doi:10.1046/j.1528-1157.2001.22001.x. PMID 11580774. http://www.blackwell-synergy.com/doi/full/10.1046/j.1528-1157.2001.22001.x.
- ^ Fisher R, van Emde Boas W, Blume W, Elger C, Genton P, Lee P, Engel J (2005). "Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE)". Epilepsia 46 (4): 470–2. doi:10.1111/j.0013-9580.2005.66104.x. PMID 15816939. http://www.blackwell-synergy.com/doi/full/10.1111/j.0013-9580.2005.66104.x.
- ^ "Epilepsy: aetiogy [sic], epidemiology and prognosis". World Health Organization. February 2001. Archived from the original on 2007-05-18. http://web.archive.org/web/20070518073641/http://www.who.int/mediacentre/factsheets/fs165/en/. Retrieved 2007-06-14.
- ^ Brodie, MJ; Elder, AT, Kwan, P (2009 Nov). "Epilepsy in later life.". Lancet neurology 8 (11): 1019–30. doi:10.1016/S1474-4422(09)70240-6. PMID 19800848.
- ^ Holmes, Thomas R. Browne, Gregory L. (2008). Handbook of epilepsy (4th ed. ed.). Philadelphia: Lippincott Williams & Wilkins. ISBN 9780781773973. http://books.google.com/books?id=gLOv8XZ5u48C&pg=PA7.
- ^ Wyllie's treatment of epilepsy : principles and practice. (5th ed. ed.). Philadelphia: Wolters Kluwer/Lippincott Williams & Wilkins. 2010. ISBN 9781582559377. http://books.google.com/books?id=mxE2FYWoY0wC&pg=PA291.
- ^ a b Cascino GD (1994). "Epilepsy: contemporary perspectives on evaluation and treatment". Mayo Clinic Proc 69: 1199–1211.
- ^ a b Engel J Jr (1996). "Surgery for seizures". NEJM 334 (10): 647–652. doi:10.1056/NEJM199603073341008. PMID 8592530.
- ^ "Proposal for revised clinical and electroencephalographic classification of epileptic seizures. From the Commission on Classification and Terminology of the International League Against Epilepsy". Epilepsia 22 (4): 489–501. 1981. doi:10.1111/j.1528-1157.1981.tb06159.x. PMID 6790275.
- ^ a b "Proposal for revised classification of epilepsies and epileptic syndromes. Commission on Classification and Terminology of the International League Against Epilepsy". Epilepsia 30 (4): 389–99. 1989. doi:10.1111/j.1528-1157.1989.tb05316.x. PMID 2502382.
- ^ Jerome Engel. "A Proposed Diagnostic Scheme For People With Epileptic Seizures And With Epilepsy: Report Of The Ilae Task Force On Classification And Terminology". ILAE. http://www.ilae-epilepsy.org/Visitors/Centre/ctf/overview.cfm. Retrieved 2006-07-18.
- ^ al., edited by Simon D. Shorvon ... et (2004). The treatment of epilepsy (2nd ed. ed.). Malden, Mass.: Blackwell Pub. ISBN 9780632060467. http://books.google.com/books?id=TfrwxdXcmosC&pg=PA3.
- ^ Olson D (November 1, 2008). "Differentiating Epileptic Seizures From Nonepileptic Spells". Consultant for Pediatricians. http://www.consultantlive.com/consultant-for-pediatricians/article/1145470/1404577.
- ^ al., edited by Simon D. Shorvon ... et (2004). The treatment of epilepsy (2nd ed.Url=http://books.google.com/books?id=TfrwxdXcmosC&pg=PA8 ed.). Malden, Mass.: Blackwell Pub. ISBN 9780632060467.
- ^ Aur D. Understanding the Physical Mechanism of Transition to Epileptic Seizures, Journal of Neuroscience Methods, Vol. 200, Issue 1, 2011, Pages 80-85 http://www.sciencedirect.com/science/article/pii/S0165027011003335
- ^ http://www.ring20.org/what-is-r20.php
- ^ Bertrand D, Picard F, Le Hellard S, Weiland S, Favre I, Phillips H, et al. (2002). "How mutations in the nAChRs can cause ADNFLE epilepsy.". Epilepsia 43 Supple 5: 112–122. doi:10.1046/j.1528-1157.43.s.5.16.x. PMID 12121305.
- ^ Loiseau P, Duche B, Cordova S, et al. (1988). "Prognosis of benign childhood epilepsy with centro-temporal spikes. A follow-up of 168 patients". Epilepsia 29 (3): 229–235. doi:10.1111/j.1528-1157.1988.tb03711.x. PMID 3371279.
- ^ Kuzniecky R (1987). "Benign occipital epilepsy: a family study.". Epilepsia 24: 346–350.
- ^ Panayiotopolous CP (2000). "Benign childhood epileptic syndromes with occipital spikes: New classification proposed by the ILAE". J Child Neurol 15 (8): 548–552. doi:10.1177/088307380001500810. PMID 10961795.
- ^ Dravet C (1978) Les epilepsies graves de l’enfant. Vie Médicale 8: 543–548
- ^ "Dravet Syndrome". http://idea-league.org/dravet-syndrome. Retrieved 2010-09-08.
- ^ (Aicardi J and Ohtahara S. Severe neonatal epilepsies with suppression-burst pattern. Epileptic Syndromes in Infancy, Childhood and Adolescence (4th edition) Eds Roger J, Bureau M, Dravet C,Genton P, Tassinari C, and Wolf P. John Libbey Eurotext 2005 ISBN 2-7420-0569-2.
- ^ Koutroumanidis M, Koepp MJ, Richardson MP, Camfield C, Agathonikou A, Ried S, et al. (1998). "The variants of reading epilepsy. A clinical and video-EEG study of 17 patients with reading-induced seizures". Brain 121 (8): 1409–1427. doi:10.1093/brain/121.8.1409. PMID 9712004.
- ^ Rogers et al.; Andrews, PI; Gahring, LC; Whisenand, T; Cauley, K; Crain, B; Hughes, TE; Heinemann, SF et al. (1994). "Autoantibodies to glutamate receptor GluR3 in Rasmussen's encephalitis". Science 265 (5172): 648–651. doi:10.1126/science.8036512. PMID 8036512.
- ^ Frucht MM, Quigg M, Schwaner C, Fountain NB. (2000). "Distribution of seizure precipitants among epilepsy syndromes". Epilepsia 41 (12): 1534–1539.. doi:10.1111/j.1499-1654.2000.001534.x. PMID 11114210.
- ^ Herzog AG, Harden CL, Liporace J, Pennell P, Schomer DL, Sperling M, et al. (2004). "Frequency of catamenial seizure exacerbation in women with localization-related epilepsy". Annals Neurology 56 (3): 431–34. doi:10.1002/ana.20214. PMID 15349872.
- ^ Harrison's Principles of Medicine. 15th edition
- ^ Miriam H. Meisler and Jennifer A. Kearney (2005). "Sodium channel mutations in epilepsy and other neurological disorders". Journal of Clinical Investigation 115 (8): 2010–2017. doi:10.1172/JCI25466. PMC 1180547. PMID 16075041. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1180547.
- ^ Shafer, Patricia Osborne; Dilorio, Colleen. "Self-Management in Epilepsy Care: Putting Teen and Families in the Center--Part One". Exceptional Parent. http://web.ebscohost.com/ehost/detail?sid=c7bbba53-243e-40f3-8352-991e2eb5b626%40sessionmgr10&vid=1&hid=12&bdata=JnNpdGU9ZWhvc3QtbGl2ZQ%3d%3d#db=eric&AN=EJ746664. Retrieved 24 October 2011.
- ^ Lopez, Jose Ignacio; Pomposo-Gaztel, Íñigo. "Patología quirúrgica de la epilepsia". Hospital Universitario de Cruces. http://www.neurologia.com/pdf/Web/5010/bd100616.pdf. Retrieved 24 October 2011.
- ^ Trost LF, Wender RC, Suter CC, Von Worley AM, Brixner DI, Rosenberg JH, Gunter MJ (December 2005). "Management of epilepsy in adults. Treatment guidelines". Postgraduate Medicine 118 (6): 29–33. PMID 16382763. http://www.postgradmed.com/index.php?art=pgm_12_2005?article=1769.
- ^ Baker GA, Jacoby A, Buck D, et al. (1997). "Quality in life in people with epilepsy: a European study". Epilepsia 38 (3): 353–362. doi:10.1111/j.1528-1157.1997.tb01128.x. PMID 9070599.
- ^ Camfiled C, Camfield P, Smith E, et al. (1986). "Asymptomatic children with epilepsy: little benefit from screening for anticonvulsant-induced liver, blood, or renal damage". Neurology 36 (6): 838–841. PMID 3703292.
- ^ mattson RH, Cramer J, COllins JF. (1985). "Comparison of carbemazipine, phenobarbital, phenytoin, and primidone in complex partial seizures". NEJM 313 (3): 145–151. doi:10.1056/NEJM198507183130303. PMID 3925335.
- ^ Kwan P, Brodie MJ. (2000). "Early identification of refractory epilepsy". NEJM 342 (5): 314–319. doi:10.1056/NEJM200002033420503. PMID 10660394.
- ^ Duncan, JS; Sander, JW, Sisodiya, SM, Walker, MC (2006 Apr 1). "Adult epilepsy.". Lancet 367 (9516): 1087–100. doi:10.1016/S0140-6736(06)68477-8. PMID 16581409.
- ^ Birbeck GL, Hays RD, Cui X, Vickrey BG. (2002). "Seizure reduction and quality of life improvements in people with epilepsy". Epilepsia 43 (5): 535–538. doi:10.1046/j.1528-1157.2002.32201.x. PMID 12027916.
- ^ Berg AT, Langfitt JT, Spencer SS, Vickrey BG. (2007). "Stopping antiepileptic drugs after epilepsy surgery: a survey of U.S. epilepsy center neurologists". Epilepsy Behav 10 (2): 219–222. doi:10.1016/j.yebeh.2006.12.001. PMC 1868701. PMID 17251061. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1868701.
- ^ Kelley K, Theodore WH (2005). "Prognosis 30 years after temporal lobectomy". Neurology 64 (11): 1974–6. doi:10.1212/01.WNL.0000163998.01543.CF. PMID 15955959.
- ^ Wiebe S, Blume WT, Girvin JP, Eliasziw M. (2001). "A randomized, controlled trial of surgery for temporal-lobe epilepsy". N Engl J Med 345 (5): 311–318. doi:10.1056/NEJM200108023450501. PMID 11484687.
- ^ al.], edited by Simon D. Shorvon ... [et (2004). The treatment of epilepsy (2nd ed. ed.). Malden, Mass.: Blackwell Pub. ISBN 9780632060467. http://books.google.com/books?id=TfrwxdXcmosC&pg=PA262.
- ^ Vogelstein, Fred (2010-11-17). "Epilepsy's Big, Fat Miracle". The New York Times. http://www.nytimes.com/2010/11/21/magazine/21Epilepsy-t.html?pagewanted=all.
- ^ Sterman M.B. (2000). "Basic concepts and clinical findings in the treatment of seizure disorders with EEG operant conditioning". Clinical Electroencephalography 31 (1): 45–55.
- ^ Theodore WH, Fisher RS (2004). "Brain stimulation for epilepsy". Lancet Neurol 3 (2): 111–118. doi:10.1016/S1474-4422(03)00664-1. PMID 14747003.
- ^ Regis, J., M. Rey, F. Bartolomei, V. Vladyka, R. Liscak, O. Schrottner and G. Pendl (2004). "Gamma knife surgery in mesial temporal lobe epilepsy: a prospective multicenter study". Epilepsia 45 (5): 504–515. doi:10.1111/j.0013-9580.2004.07903.x. PMID 15101832.
- ^ Takahashi, T. and Y. Tsukahara (1992). "Usefulness of blue sunglasses in photosensitive epilepsy". Epilepsia 33 (3): 517–521. doi:10.1111/j.1528-1157.1992.tb01702.x. PMID 1592030.
- ^ Barriaux, Marianne (2006-10-16). "Dogs trained to warn of an imminent epileptic fit". London: The Guardian. http://business.guardian.co.uk/story/0,,1923146,00.html. Retrieved 2006-11-24.
- ^ Cheuk D, Wong V (2006). Cheuk, Daniel. ed. "Acupuncture for epilepsy". Cochrane Database Syst Rev (2): CD005062. doi:10.1002/14651858.CD005062.pub2. PMID 16625622.
- ^ Ramaratnam S, Baker GA, Goldstein LH (2005). Ramaratnam, Sridharan. ed. "Psychological treatments for epilepsy". Cochrane Database Syst Rev (4): CD002029. doi:10.1002/14651858.CD002029.pub2. PMID 16235293.
- ^ Ranganathan LN, Ramaratnam S (2005). Ranganathan, Lakshmi Narasimhan. ed. "Vitamins for epilepsy". Cochrane Database Syst Rev (2): CD004304. doi:10.1002/14651858.CD004304.pub2. PMID 15846704.
- ^ Ramaratnam S, Sridharan K (2000). Ramaratnam, Sridharan. ed. "Yoga for epilepsy". Cochrane Database Syst Rev (3): CD001524. doi:10.1002/14651858.CD001524. PMID 10908505.
- ^ Arida RM, Scorza FA, Scorza CA, Cavalheiro EA (2009). "Is physical activity beneficial for recovery in temporal lobe epilepsy? Evidences from animal studies". Neurosci Biobehav Rev (33): 422–431.
- ^ Arida RM, Cavalheiro EA, da Silva AC, Scorza FA. (2008). "Physical activity and epilepsy: proven and predicted benefits". Sports Med. (38(7)): 607–615.
- ^ Hirtz D, Thurman DJ, Gwinn-Hardy K, Mohamed M, Chaudhuri AR, Zalutsky R (2007-01-30). "How common are the 'common' neurologic disorders?". Neurology 68 (5): 326–37. doi:10.1212/01.wnl.0000252807.38124.a3. PMID 17261678.
- ^ Hauser, WA, Kurland, LT (1975). "The epidemiology of epilepsy in Rochester, Minnesota, 1935 through 1967". Epilepsia 16 (1): 1–66. doi:10.1111/j.1528-1157.1975.tb04721.x. PMID 804401.
- ^ Sander JW (2003). "The epidemiology of epilepsy revisited". Curr Opin Neurol 16 (2): 165–70. doi:10.1097/00019052-200304000-00008. PMID 12644744.
- ^ Walczak TS, Leppik IE, D'Amelio M, Rarick J, So E, Ahman P, Ruggles K, Cascino GD, Annegers JF, Hauser WA (2001). "SIncidence and risk factors in sudden unexpected death in epilepsy: a prospective cohort study". Neurology 56 (4): 519–525. PMID 11222798.
- ^ Lathers, C. and P. Schraeder (1990). Epilepsy and Sudden Death. Dekker, NY, NY.
- ^ Hitiris, N., R. Mohanraj, J. Norrie and M. J. Brodie (2007). "Mortality in epilepsy". Epilepsy Behavior 10 (3): 363–376. doi:10.1016/j.yebeh.2007.01.005. PMID 17337248.
- ^ Hanna et al 2002
- ^ Hanna et al, (2002) The National Sentinel Audit of Epilepsy Related Death, The Stationary Office, London.
- ^ Plioplys S, Dunn DW, Caplan R (2007). "10-year research update review: psychiatric problems in children with epilepsy". J Am Acad Child Adolesc Psychiatry 46 (11): 1389–402. doi:10.1097/chi.0b013e31815597fc. PMID 18049289.
- ^ Reilly, Colin J.. "Attention Deficit Hyperactivity Disorder (ADHD) in Childhood Epilepsy". Research in Developmental Disabilities: A Multidisciplinary Journal. http://web.ebscohost.com/ehost/detail?sid=bc023b45-4a9e-4944-ac22-82331397675e%40sessionmgr13&vid=1&hid=12&bdata=JnNpdGU9ZWhvc3QtbGl2ZQ%3d%3d#db=eric&AN=EJ918684. Retrieved 24 October 2011.
- ^ Levisohn PM (2007). "The autism-epilepsy connection". Epilepsia 48 (Suppl 9): 33–5. doi:10.1111/j.1528-1167.2007.01399.x. PMID 18047599.
- ^ OED. Retrieved 8 September 2009.
- ^ a b c "Epilepsy: historical overview". Health Topics A TO Z. http://allcountries.org/health/epilepsy_historical_overview.html. Retrieved 2011-03-20.
- ^ When Epilepsy Goes By Another Name | epilepsy.com
- ^ Jilek-Aall, L (1999). "Morbus sacer in Africa: some religious aspects of epilepsy in traditional cultures". Epilepsia 40 (3): 382–6. doi:10.1111/j.1528-1157.1999.tb00723.x. PMID 10080524.
- ^ Hippocrates quotes
- ^ Epilepsy Foundation Driving and You - Can you drive an automobile if you have epilepsy?.
- ^ Epilepsy Foundation Driver Information by State
- ^ UK Epilepsy Action: Driving and Epilepsy, I've had a seizure. What should I do?
- ^ UK Driver and Vehicle Licensing Agency Guide to the Current Medical Standards Of Fitness to Drive. Full details for doctors regarding epilepsy are given in the Appendix. Information for drivers can be found in Medical Rules - Group 1 Licence Holders
- ^ UK Epilepsy Action: booklet with further details about driving PDF
- ^ Epilepsy Action (2009), Driving law relating to seizures. Available from http://www.epilepsy.org.uk/info/driving/lawseizure (Accessed on 15 February 2009)
- ^ United States Code of Federal Regulations, Title 14, sections 67.109(a)(1), 67.209(a)(1), 67.309(a)(1)
- ^ FAA Aerospace Medical Dispositions, Item 46: Neurologic, [1], retrieved 20 December 2010
- ^ Civil Aviation Authority requirements - NPPL[2], retrieved 06 May 2011
- ^ a b Thomas, WB (2010 Jan). "Idiopathic epilepsy in dogs and cats.". The Veterinary clinics of North America. Small animal practice 40 (1): 161–79. doi:10.1016/j.cvsm.2009.09.004. PMID 19942062.
Further reading
- Morrow, Jim. 2011. Epilepsy a patient's handbook. National Services for Health Improvement. ISBN 978-0-9560921-7-5.
- Walker, M. & Shorvon, S. Understanding Epilepsy second edition 2000. Family Doctor Pulications. ISBN 1 898205 20 5.
Seizures and epilepsy (G40–G41, 345) Basics Seizure types · Seizure trigger · Breakthrough seizure · Postictal state · Epileptogenesis · Seizure prediction · Aura (warning sign)Treatments Antiepileptics · Template:Anticonvulsants (for list) · Electroencephalography (diagnosis method) · EpileptologistRelated disorders Epilepsy organizations Epilepsy Foundation (USA) · International Dravet Epilepsy Action League · Epilepsy Toronto · Epilepsy Research UK · Epilepsy Action Australia · Citizens United for Research in Epilepsy · Epilepsy Action · Epilepsy SocietyIssues for epileptics Seizure types
Epilepsy typesPartial/focal Seizures: Simple partial · Complex partial · Jacksonian seizure
Epilepsy: Temporal lobe epilepsy · Frontal lobe epilepsy · Rolandic epilepsy · Nocturnal epilepsyGeneralised Tonic-clonic · Absence seizure · Atonic seizure · Automatism · Benign familial neonatal · Lennox-Gastaut · WestStatus epilepticus Myoclonic epilepsy Non-epileptic seizures Categories:- Medical terms
- Epilepsy
- Greek loanwords
- Neurological disorders in children
- Disorders causing seizures

Wikimedia Foundation. 2010.