- Tuberous sclerosis
Infobox_Disease
Name = Tuberous sclerosis
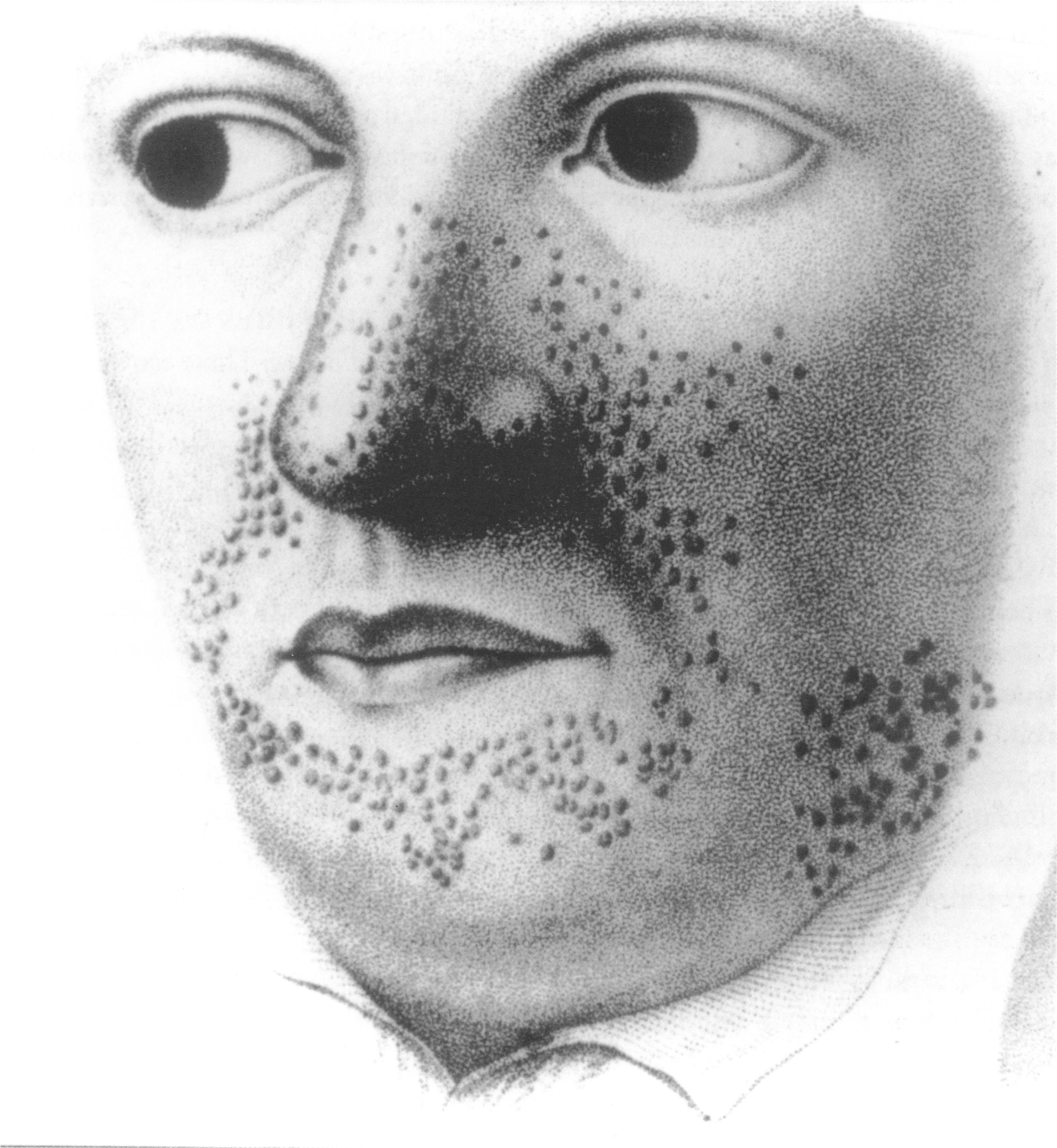
Caption = Earliest illustration, from Rayer's atlas of skin diseases, 1835.
DiseasesDB = 13433
ICD10 = ICD10|Q|85|1|q|80
ICD9 = ICD9|759.5
ICDO =
OMIM = 191100
MedlinePlus = 000787
eMedicineSubj = neuro
eMedicineTopic = 386
eMedicine_mult = eMedicine2|derm|438 eMedicine2|ped|2796 eMedicine2|radio|723
MeshID = D014402Tuberous sclerosis or tuberous sclerosis complex (TSC) is a rare, multi-system genetic disease that causes benign tumours to grow in thebrain and on other vital organs such as thekidney s,heart ,eye s,lung s, andskin . A combination of symptoms may includeseizure s, developmental delay, behavioural problems, skin abnormalities, lung and kidney disease. TSC is caused bymutation s on either of twogene s,TSC1 andTSC2 , which encode for theprotein s hamartin and tuberin respectively. These proteins act as tumour growth suppressors, agents that regulate cell proliferation and differentiation.cite web
url = http://www.ninds.nih.gov/disorders/tuberous_sclerosis/detail_tuberous_sclerosis.htm
title = Tuberous Sclerosis Fact Sheet
accessdate = 2007-01-09
date = 2006-04-11
publisher = NINDS (Some text copied with permission.)]The name, composed of the Latin "tuber" (swelling) and the Greek "skleros" (hard), refers to the
pathological finding of thick, firm and pale gyri, called "tubers", in the brains of patientspostmortem . These tubers were first described byDésiré-Magloire Bourneville in 1880; the cortical manifestations may sometimes still be known by theeponym Bourneville's disease.igns and symptoms
The physical manifestations of tuberous sclerosis are due to the formation of
hamartia (malformed tissue such as the cortical tubers),hamartoma s (benign growths such as facial angiofibroma and subependymal nodules) and, very rarely, canceroushamartoblastoma s. The effect of these on the brain leads to neurological symptoms such asseizure s, developmental delay and behavioral problems.Central nervous system
About 50% of people with TSC have
learning difficulties ranging from mild to profound,cite journal
author = Ridler K, "et al"
title = Neuroanatomical Correlates of Memory Deficits in Tuberous Sclerosis Complex
journal = Cerebral Cortex
year = 2006
pmid = 16603714 | volume = 17
pages = 261
doi = 10.1093/cercor/bhj144] and studies have reported that between 25% and 61% of affected individuals meet the diagnostic criteria forautism , with an even higher proportion showing features of a broaderpervasive developmental disorder .cite journal
author = Harrison JE, Bolton, PF
title = Annotation: Tuberous sclerosis
journal = Journal of Child Psychology and Psychiatry
year = 1997
pages = 603–614
volume = 38
pmid = 9315970| doi = 10.1111/j.1469-7610.1997.tb01687.x] A 2008 study reportedself-injurious behavior in 10% of people with TSC. [cite journal |journal= Epilepsy Behav |year=2008 |title= Self-injurious behavior and tuberous sclerosis complex: frequency and possible associations in a population of 257 patients |author= Staley BA, Montenegro MA, Major P "et al." |pmid=18703161] Other conditions, such asADHD , aggression, behavioral outbursts andOCD can also occur. Lower IQ is associated with more brain involvement on MRI.Three type of brain tumours may be associated with TSC:i. Giant cell astrocytoma: (grows and blocks the CSF flowleading to dilatation of ventricles causing headacheand vomiting.ii. Cortical tubers: after which the disease is named.iii. Sub-ependymal nodules: form in the walls of ventricles.
Classic intracranial manifestations of tuberous sclerosis include sub
ependyma l nodules and cortical/subcortical tubers. cite journal
author = Ridler K, "et al"
title = Standardized whole brain mapping of tubers and subependymal nodules in tuberous sclerosis complex
journal = Journal of Child Neurology
year = 2004
volume = 19
issue = 9
pages = 658–665
pmid = 15563011]The tubers are typically triangular in configuration, with the apex pointed towards the ventricles, and are thought to represent foci of abnormal neuronal migration. The T2 signal abnormalities may subside in adulthood, but will still be visible on histopathological analysis. On magnetic resonance imaging, TSC patients can exhibit other signs consistent with abnormal neuron migration (radial white matter tracts hyperintense on T2WI, heterotopic gray matter).
Subependymal nodules are composed of abnormal, swollen glial cells and bizarre multinucleated cells which are indeterminate for glial or neuronal origin. There is no interposed neural tissue. These nodules have a tendency to calcify as the patient ages. A nodule that markedly enhances and enlarges over time should be considered suspicious for transformation into a subependymal giant cell astrocytoma (SEGA). A SEGA typically develops in the region of the foramen of Monro, in which case it is at risk of developing an obstructive
hydrocephalus .A variable degree of ventricular enlargement, either obstructive (e.g. by a subependymal nodule in the region of the foramen of Monroe) or idiopathic in nature.
Kidneys
Between 60 and 80% of TSC patients have benign tumors (hamartomas) of the kidneys called
angiomyolipoma s (AML) frequently causinghematuria . These tumors are composed of vascular tissue ("angio–"),smooth muscle ("–myo–"), andfat ("–lipoma "). Although benign, an AML larger than 4 cm is at risk for a potentially catastrophic hemorrhage either spontaneously or with minimal trauma. AMLs are found in about 1 in 300 people without TSC. However those are usually solitary, whereas in TSC they are commonly multiple and bilateral.Approximately 20-30% of people with TSC will have renal cysts, causing few problems. However, 2% may also have
autosomal dominant polycystic kidney disease .Very rare (< 1%) problems include
renal cell carcinoma andoncocytoma s (benign adenomatous hamartoma ).Lungs
Patients with TSC can develop progressive replacement of the lung parenchyma with multiple cysts. This process is identical to another disease called
lymphangioleiomyomatosis (LAM). Recent genetic analysis has shown that the proliferative bronchiolar smooth muscle in tuberous sclerosis-related LAM is monoclonal metastasis from a coexisting renal angiomyolipoma. There have been cases of TSC-related LAM recurring following lung transplant. cite journal
author = Henske EP.
title = Metastasis of benign tumor cells in tuberous sclerosis complex
journal = Genes, Chromosomes & Cancer
year = 2003
volume = 38
issue = 4
pages = 376–381
pmid = 14566858| doi = 10.1002/gcc.10252]Heart
Rhabdomyoma s are benign tumors of striated muscle. A cardiac rhabdomyoma can be discovered usingechocardiography in approximately 50% of people with TSC. However the incidence in the newborn may be as high as 90% and in adults as low as 20%. These tumors grow during the second half of pregnancy and regress after birth. Many will disappear entirely. Alternatively, the tumor size remains constant as the heart grows, which has much the same effect.Problems due to rhabdomyomas include obstruction, arrhythmia and a murmur. Such complications occur almost exclusively during pregnancy or within the child's first year.
Prenatal ultrasound, performed by an obstetric sonographer specializing in cardiology, can detect a rhabdomyoma after 20 weeks. This rare tumour is a strong indicator of TSC in the child, especially if there is a family history of TSC.
kin
Some form of dermatological sign will be present in 96% of individuals with TSC. Most cause no problems but are helpful in diagnosis. Some cases may cause disfigurement, necessitating treatment. The most common skin abnormalities include:
* Facial angiofibromas ("adenoma sebaceum"): A rash of reddish spots or bumps, which appear on the nose and cheeks in a butterfly distribution. They consist of blood vessels and fibrous tissue. This socially embarrassing rash starts to appear during childhood and can be removed using
dermabrasion or laser treatment.
* Ungual or subungual fibromas"': Small fleshy tumors that grow around and under the toenails or fingernails and may need to be surgically removed if they enlarge or cause bleeding. These are very rare in childhood but common by middle age.
* Hypomelanicmacules ("ash leaf spots"): White or lighter patches of skin that may appear anywhere on the body and are caused by a lack ofmelanin . These are usually the only visible sign of TSC at birth. In fair-skinned individuals aWood's lamp (ultraviolet light) may be required to see them.
* Forehead plaques: Raised, discolored areas on the forehead.
* Shagreen patches: Areas of thick leathery skin that are dimpled like an orange peel, usually found on the lower back or nape of the neck.
* Other skin features are not unique to individuals with TSC, including molluscum fibrosum or skin tags, which typically occur across the back of the neck and shoulders, "café au lait" spots or flat brown marks, and poliosis, a tuft or patch of white hair on the scalp or eyelids.Eyes
Retinal lesions, called astrocytic hamartomas, which appear as a greyish or yellowish-white lesion in the back of the globe on the ophthalmic examination. Astrocytic hamartomas can calcify, and in is in the differential diagnosis of a calcified globe mass on a CT scan.
Non-retinal lesions associated with TSC include
*Coloboma
*Angiofibromas of the eyelids
*Papilledema (related to hydrocephalus)Variability
Individuals with tuberous sclerosis may experience none or all of the clinical signs discussed above. The following table shows the prevalence of some of the clinical signs in individuals diagnosed with tuberous sclerosis.
Genetics
Tuberous sclerosis is a genetic disorder with an
autosomal dominant pattern of inheritance, andpenetrance is 100%. [cite web
url = http://www.geneclinics.org/profiles/tuberous-sclerosis/details.html
title = Tuberous Sclerosis Complex
accessdate = 2007-09-02
author = Northrup H, Au K
date =5 December 2005
work = GeneReviews] Two thirds of TSC cases result from sporadic genetic mutations, not inheritance, but their offspring may inherit it from them. Current genetic tests have difficulty locating the mutation in approximately 20% of individuals diagnosed with the disease. So far it has been mapped to two genetic loci, TSC1 andTSC2 .TSC1 encodes for the protein
hamartin , is located onchromosome 9 q34 and was discovered in 1997.cite journal
author = van Slegtenhorst M, de Hoogt R, Hermans C, "et al"
title = Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34
journal = Science
volume = 277
issue = 5327
pages = 805–8
year = 1997
pmid = 9242607
doi = 10.1126/science.277.5327.805] TSC2 encodes for the protein tuberin, is located onchromosome 16 p13.3 and was discovered in 1993.cite journal
author = European Chromosome 16 Tuberous Sclerosis Consortium
title = Identification and characterization of the tuberous sclerosis gene on chromosome 16
journal = Cell
volume = 75
issue = 7
pages = 1305–15
year = 1993
pmid = 8269512] TSC2 is contiguous with PKD1, the gene involved in one form ofpolycystic kidney disease (PKD). Gross deletions affecting both genes may account for the 2% of individuals with TSC who also develop PKD in childhood.cite journal
author=Brook-Carter PT, "et al"
title=Deletion of the TSC2 and PKD1 genes associated with severe infantile polycystic kidney disease--a contiguous gene syndrome
journal = Nature Genetics
year = 1994
pages = 328–32
volume = 8
issue = 4
pmid = 7894481| doi=10.1038/ng1294-328] TSC2 has been associated with a more severe form of TSC.cite journal
author=Dabora SL, "et al"
title=Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs
journal = American Journal of Human Genetics
year = 2001
pages = 64–80
volume = 68
issue = 1
url = http://www.pubmedcentral.nih.gov/picrender.fcgi?artid=1234935&blobtype=pdf
pmid = 11112665| doi=10.1086/316951] However, the difference is subtle and cannot be used to identify the mutation clinically. Estimates of the proportion of TSC caused by TSC2 range from 55% to 80-90%.cite journal
author = Rendtorff ND, "et al"
title = Analysis of 65 tuberous sclerosis complex (TSC) patients by TSC2 DGGE, TSC1/TSC2 MLPA, and TSC1 long-range PCR sequencing, and report of 28 novel mutations
journal = Human Mutation
year = 2005
pages = 374–83
volume = 26
issue = 4
pmid = 16114042
doi = 10.1002/humu.20227]TSC1 and TSC2 are both
tumor suppressor gene s that function according to Knudson's "two hit" hypothesis. That is, a second random mutation must occur before a tumor can develop. This explains why, despite its 100 percentpenetrance , TSC has wideexpressivity .protein
Name = Hamartin
caption =
width =
HGNCid = 12362
Symbol = TSC1
AltSymbols =
EntrezGene = 7248
OMIM = 605284
RefSeq = NM_000368
UniProt = Q92574
PDB =
ECnumber =
Chromosome = 9
Arm = q
Band = 34
LocusSupplementaryData =protein
Name = Tuberin
caption =
width =
HGNCid = 12363
Symbol =TSC2
AltSymbols =
EntrezGene = 7249
OMIM = 191092
RefSeq = NM_000548
UniProt = P49815
PDB =
ECnumber =
Chromosome = 16
Arm = p
Band = 13.3
LocusSupplementaryData =Pathophysiology
Hamartin and tuberin function as a complex which is involved in the control of cell growth and cell division. (The complex appears to be a
Rheb GTPase which suppresses mTOR signalling, part of thegrowth factor (insulin ) signalling pathway.) Thus, mutations at the TSC1 and TSC2 loci result in a loss of control of cell growth and cell division, and therefore a predisposition to forming tumors.Diagnosis
There are no
pathognomonic clinical signs for tuberous sclerosis. Many signs are present in individuals who are healthy (although rarely), or who have another disease. A combination of signs, classified as major or minor, is required in order to establish a clinical diagnosis.In infants, the first clue is often the presence of seizures, delayed development or white patches on the skin. A full clinical diagnosis involvescite web
url = http://www.ninds.nih.gov/disorders/tuberous_sclerosis/detail_tuberous_sclerosis.htm
title = Tuberous Sclerosis Fact Sheet
accessdate = 2006-10-03
date =2006-04-11
publisher = National Institute of Neurological Disorders and Stroke] cite web
url = http://www.tuberous-sclerosis.org/publications/clinicalguidelinessummary.pdf
title = Summary of Clinical guidelines for the care of patients with Tuberous Sclerosis Complex
accessdate = 2006-10-03
year = 2002
month = April
publisher = Tuberous Sclerosis Association]
* Taking a personal and family history.
* Examining the skin under aWood's lamp (hypomelanotic macules), the fingers and toes (ungual fibroma), the face (angiofibromas) and the mouth (dental pits and gingival fibromas).
* Cranial imaging with non enhanced CT or, preferably,MRI (cortical tubers and subependymal nodules).
* Renalultrasound (angiomyolipoma or cysts).
* Anechocardiogram in infants (rhabdomyoma).
*Fundoscopy (retinal nodular hamartomas or achromic patch).The various signs are then marked against the diagnostic criteria to produce a level of diagnostic certainty:*Definite – Either two major features or one major feature plus two minor features.
*Probable – One major plus one minor feature.
*Suspect – Either one major feature or two or more minor features.Due to the wide variety of mutations leading to TSC, there are no simple genetic tests available to identify new cases. Nor are there any biochemical markers for the gene defects. However, once a person has been clinically diagnosed, the genetic mutation can usually be found. The search is time-consuming and has a 15% failure rate, which is thought to be due to
somatic mosaicism. If successful, this information can be used to identify affected family members, includingprenatal diagnosis .As of 2006 ,preimplantation diagnosis is not widely available.Management
Drug therapy for some of the manifestations of TSC is currently in the developmental stage. [Yates JR. "Tuberous sclerosis". Eur J Hum Genet. 2006 Jul 26; PMID 16868562 ] For example, a 2008 study found that treatment with
rapamycin rescued learning and memory deficits in a mouse model of tuberous sclerosis. [cite journal |journal= Nat Med |date=2008 |title= Reversal of learning deficits in a "Tsc2"+/- mouse model of tuberous sclerosis |author= Ehninger D, Han S, Shilyansky C "et al." |doi=10.1038/nm1788 |pmid=18568033 |laysummary=http://www.sciencedaily.com/releases/2008/06/080622224428.htm |laysource= Science News |laydate=2008-06-23]Community TSC is a distributed computing project to find drugs to treat TSC.Fact|date=June 2008|laysummary=http://www.childhooddiseases.org/community_tsc.htmlThe patients usually have relapse of symptoms in theclinical course. Unless any vital function is affected, lifeexpectancy is good. Majority of patients will require somemedications to control symptoms, e.g., anti-epileptics tocontrol seizures.Prognosis
The prognosis for individuals with TSC depends on the severity of symptoms, which range from mild skin abnormalities to varying degrees of learning disabilities and epilepsy to severe mental retardation, uncontrollable seizures, and kidney failure. Those individuals with mild symptoms generally do well and live long productive lives, while individuals with the more severe form may have serious disabilities. However, with appropriate medical care, most individuals with the disorder can look forward to normal life expectancy.
Leading causes of death include renal disease, brain tumour, lymphangiomyomatosis of the lung, and status epilepticus or bronchopneumonia in those with severe mental handicap.cite journal
author = Shepherd C, Gomez M, Lie J, Crowson C
title = Causes of death in patients with tuberous sclerosis
journal = Mayo Clin Proc
volume = 66
issue = 8
pages = 792–6
year = 1991
pmid = 1861550] Cardiac failure due to rhabdomyomas is a risk in the fetus or neonate, but is rarely a problem subsequently. Kidney complications such as angiomyolipoma (AML) and cysts are common, and more frequent in females than males and in TSC2 than TSC1. Renal cell carcinoma is uncommon. Lymphangioleiomyomatosis (LAM) is only a risk for females with AMLs.cite journal
author = Rakowski SK, Winterkorn EB, Paul E, Steele DJ, Halpern EF, Thiele EA.
title = Renal manifestations of tuberous sclerosis complex: Incidence, prognosis, and predictive factors
journal = Kidney International
volume = 70
issue =
pages = 1777
year = 2006
pmid = 17003820
doi = 10.1038/sj.ki.5001853] In the brain, the subependymal nodules occasionally degenerate to subependymal giant cell astrocytomas (SEGA). These may block the circulation of cerebrospinal fluid around the brain, leading to hydrocephalus.Detection of the disease should prompt one for geneticcounselling. It is also important to know that even thoughthe disease does not have a cure, symptoms can be treatedsymptomatically. Hence, awareness regarding differentorgan manifestations of tuberous sclerosis is important.
Epidemiology
Tuberous sclerosis occurs in all races and ethnic groups, and in both genders. The live-birth
prevalence is estimated to be between 10 and 16 cases per 100,000. A 1998 study estimated total population prevalence between about 7 and 12 cases per 100,000, with more than half of these cases undetected.cite journal
author = O'Callaghan FJK, Shiell AW, Osborne JP, Martyn CN
title = Prevalence of tuberous sclerosis estimated by capture-recapture analysis
journal = Lancet
volume = 351
issue = 9114
year = 1998
pages = 1490
doi = 10.1016/S0140-6736(05)78872-3] These estimates are significantly higher than those produced by older studies, when tuberous sclerosis was regarded as an extremely rare disease. The reason is that the invention of CT and ultrasound scanning have enabled the diagnosis of many non-symptomatic cases. Prior to this, the diagnosis of tuberous sclerosis was largely restricted to severely affected individuals with Vogt's triad of learning disability, seizures and facial angiofibroma. The total population prevalence figures have steadily increased from 1:150,000 in 1956, to 1:100,000 in 1968, to 1:70,000 in 1971, to 1:34,200 in 1984, to the present figure of 1:12,500 in 1998. Whilst still regarded as arare disease , it is common when compared to many other genetic diseases.History
Tuberous sclerosis first came to medical attention when dermatologists described the distinctive facial rash (1835 and 1850). A more complete case was presented by von Recklinghausen (1862) who identified heart and brain tumours in a newborn that had only briefly lived. However, Bourneville (1880) is credited with having first characterized the disease, coining the name "tuberous sclerosis", thus earning the
eponym Bourneville's disease. The neurologist Vogt (1908) established a diagnostic triad of epilepsy, idiocy, and adenoma sebaceum (an obsolete term for facial angiofibroma).Curatolo (2003), chapter: "Historical Background".]Symptoms were periodically added to the clinical picture. The disease as presently understood was first fully described by Gomez (1979). The invention of medical ultrasound, CT and MRI has allowed physicians to examine the internal organs of live patients and greatly improved diagnostic ability.
Two genetic loci associated with tuberous sclerosis, TSC1 and
TSC2 , were discovered in 1997 and 1992 respectively. This has enabled the use of genetic testing as a diagnostic tool. The proteins associated with TSC1 and TSC2, harmartin and tuberin, function as a complex in the mTOR signalling pathway that controls cell growth and cell division. The importance of this pathway in cancer therapy has stimulated further research into Tuberous Sclerosis.In 2002, treatment with
rapamycin was found to be effective at shrinking tumours in animals. This has led to human trials of rapamycin as a drug to treat several of the tumors associated with Tuberous Sclerosis.cite web
url = http://www.tsdev.de/92001/Uploaded/hhehn%7Cgeschichte_der_tsc2005.pdf
title = Zur Geschichte der Tuberösen Sklerose (The History of Tuberous Sclerosis)
accessdate = 2007-01-08
author = Rott HD, Mayer K, Walther B, Wienecke R
year = 2005
month = 03
publisher = Tuberöse Sklerose Deutschland e.V
language = German]Notes
References
*Ingole J; Jain
*cite book | author=Paolo Curatolo (Editor) | title=Tuberous Sclerosis Complex : From Basic Science to Clinical Phenotypes | publisher=MacKeith Press | year=2003 | isbn = 1-898683-39-5
*External links
* The HUGO Gene Nomenclature Committee web pages for [http://www.gene.ucl.ac.uk/nomenclature/data/get_data.php?hgnc_id=HGNC:12362 TSC1] and [http://www.gene.ucl.ac.uk/nomenclature/data/get_data.php?hgnc_id=HGNC:12363 TSC2] provide a selection of links for further genetic research.
* The [http://www.childhooddiseases.org/ Rothberg Institute For Childhood Diseases] have adistributed computing project calledCommunityTSC
* [http://dermatlas.med.jhmi.edu/derm/result.cfm?Diagnosis=189 Dermatlas: Images of Tuberous sclerosis]
* Mass General Hospital's [http://www.massgeneral.org/livingwithtsc/ Living with TSC]upport groups
* United Kingdom: [http://www.tuberous-sclerosis.org/ The Tuberous Sclerosis Association] . Awareness month is October.
* United States: [http://www.tsalliance.org/ Tuberous Sclerosis Alliance] . Awareness month is May.
* Canada: [http://www.tscanada.ca/ Tuberous Sclerosis Canada] . Awareness month is May.
* Australasia: [http://www.atss.org.au/ Australasian Tuberous Sclerosis Society] .
* Brazil: [http://www.abet.org.br/entrada.htm Associação Brasileira de Esclerose Tuberosa ("Brazilian Tuberous Sclerosis Association")] pt icon
* Taiwan: [http://www.ttsc.org.tw Taiwan Tuberous Sclerosis Complex]
:_79-81_)
Wikimedia Foundation. 2010.