- Amyotrophic lateral sclerosis
-
This article is about progressive Motor Neuron Disease (MND) affecting both the upper motor neurons and lower motor neurons. For MND affecting either but not necessarily both, see Motor neurone disease."ALS" redirects here. For other uses, see ALS (disambiguation).
Amyotrophic Lateral Sclerosis
(Lou Gehrig's Disease)Classification and external resources 
This MRI (parasagittal FLAIR) demonstrates increased T2 signal within the posterior part of the internal capsule and can be tracked to the subcortical white matter of the motor cortex, outlining the corticospinal tract, consistent with the clinical diagnosis of ALS.ICD-10 G12.2 ICD-9 335.20 OMIM 105400 DiseasesDB 29148 MedlinePlus 000688 eMedicine neuro/14 emerg/24 pmr/10 MeSH D000690 Amyotrophic lateral sclerosis (ALS), also referred to as Lou Gehrig's disease, is a form of motor neuron disease caused by the degeneration of neurons located in the ventral horn of the spinal cord and the cortical neurons that provide their efferent input. The condition is often called Lou Gehrig's disease in North America, after the New York Yankees baseball player who was diagnosed with the disease in 1939. The disorder is characterized by rapidly progressive weakness, muscle atrophy and fasciculations, spasticity, dysarthria, dysphagia, and respiratory compromise. Sensory function generally is spared, as is autonomic, and oculomotor activity. ALS is a progressive,[1] fatal, neurodegenerative disease with most affected patients dying of respiratory compromise and pneumonia after 2 to 3 years; although some perish within a year from the onset of symptoms, and occasional individuals have a more indolent course and survive for many years.[2]
Contents
Signs and symptoms
The disorder causes muscle weakness and atrophy throughout the body caused by degeneration of the upper and lower motor neurons. Unable to function, the muscles weaken and atrophy. Affected individuals may ultimately lose the ability to initiate and control all voluntary movement, although bladder and bowel sphincters and the muscles responsible for eye movement are usually, but not always, spared.
Cognitive function is generally spared for most patients although some (~5%) also have frontotemporal dementia.[3] A higher proportion of patients (~30-50%) also have more subtle cognitive changes which may go unnoticed but are revealed by detailed neuropsychological testing. Sensory nerves and the autonomic nervous system, which controls functions like sweating, are generally unaffected but may be involved for some patients.
Initial symptoms
The earliest symptoms of ALS are typically obvious weakness and/or muscle atrophy. Other presenting symptoms include muscle fasciculation (twitching), cramping, or stiffness of affected muscles; muscle weakness affecting an arm or a leg; and/or slurred and nasal speech. The parts of the body affected by early symptoms of ALS depend on which motor neurons in the body are damaged first. About 75% of people contracting the disease experience "limb onset" ALS i.e. first symptoms in the arms ("upper limb", not to be confused with "upper motor neuron") or legs ("lower limb", not to be confused with "lower motor neuron"). Patients with the leg onset form may experience awkwardness when walking or running or notice that they are tripping or stumbling, often with a "dropped foot" which drags gently along the ground. Arm-onset patients may experience difficulty with tasks requiring manual dexterity such as buttoning a shirt, writing, or turning a key in a lock. Occasionally, the symptoms remain confined to one limb for a long period of time or for the whole length of the illness; this is known as monomelic amyotrophy.
About 25% of cases are "bulbar onset" ALS. These patients first notice difficulty speaking clearly or swallowing. Speech may become slurred, nasal in character, or quieter. Other symptoms include difficulty swallowing, and loss of tongue mobility. A smaller proportion of patients experience "respiratory onset" ALS where the intercostal muscles that support breathing are affected first.
Regardless of the part of the body first affected by the disease, muscle weakness and atrophy spread to other parts of the body as the disease progresses. Patients experience increasing difficulty moving, swallowing (dysphagia), and speaking or forming words (dysarthria). Symptoms of upper motor neuron involvement include tight and stiff muscles (spasticity) and exaggerated reflexes (hyperreflexia) including an overactive gag reflex. An abnormal reflex commonly called Babinski's sign (the big toe extends upward and other toes spread out) also indicates upper motor neuron damage. Symptoms of lower motor neuron degeneration include muscle weakness and atrophy, muscle cramps, and fleeting twitches of muscles that can be seen under the skin (fasciculations). Around 15–45% of patients experience pseudobulbar affect, also known as "emotional lability", which consists of uncontrollable laughter, crying or smiling, attributable to degeneration of bulbar upper motor neurons resulting in exaggeration of motor expressions of emotion.
To be diagnosed with ALS, patients must have signs and symptoms of both upper and lower motor neuron damage that cannot be attributed to other causes.
Disease progression
Although the sequence of emerging symptoms and the rate of disease progression vary from person to person, eventually most patients are not able to stand or walk, get in or out of bed on their own, or use their hands and arms. Difficulty swallowing and chewing impair the patient's ability to eat normally and increase the risk of choking or aspirating food/liquids into the lungs. Aspiration pneumonia and weight maintenance can then become a problem. Because the disease usually does not affect cognitive abilities, patients are aware of their progressive loss of function and may become anxious and depressed. A small percentage of patients go on to develop frontotemporal dementia characterized by profound personality changes; this is more common among those with a family history of dementia. A larger proportion of patients experience mild problems with word-generation, attention, or decision-making. Cognitive function may be affected as part of the disease process or could be related to poor breathing at night (nocturnal hypoventilation). Health care professionals need to explain the course of the disease and describe available treatment options so that patients can make informed decisions in advance.
As the diaphragm and intercostal muscles (rib cage) weaken, forced vital capacity and inspiratory pressure diminish. In bulbar onset ALS, this may occur before significant limb weakness is apparent. Bilevel positive pressure ventilation (frequently referred to by the tradename BiPAP) is frequently used to support breathing, first at night, and later during the daytime as well. It is recommended that long before BiPAP becomes insufficient, patients must decide whether to have a tracheostomy and long term mechanical ventilation. At this point, some patients choose palliative hospice care. Most people with ALS die of respiratory failure or pneumonia. Death usually occurs within two to five years of diagnosis. Although the disease can strike at any age, most people are between forty and seventy years of age when the disease strikes and men are affected slightly more frequently than women. An estimated 5,000 people in the United States are diagnosed with the disease each year.[4] ALS, a progressive disease, leads to death in half of the people diagnosed within three years and ninety percent within six years. In a population based study in Minnesota, USA, looking back over 85 years, 14 percent of people with ALS survived more than 5 years. Those who survived 5 years or longer were clinically similar to the total population ALS population in terms of gender, age, gender, and site of onset, but they had a longer time from symptomatic onset to time of diagnosis [5]
ALS predominantly affects the motor neurons, and in the majority of cases the disease does not impair a patient's mind, personality, intelligence, or memory. Nor does it affect a person's ability to see, smell, taste, hear, or feel touch. Control of eye muscles is the most preserved function, although some patients with an extremely long duration of disease (20+ years) may lose eye control too. Unlike multiple sclerosis, bladder and bowel control are usually preserved in ALS, although as a result of immobility and diet changes, intestinal problems such as constipation can require intensive management.
Cause
For patients without a family history of the disease, which includes ~95% of cases, there is no known cause for ALS.
There is a known hereditary factor in familial ALS (FALS), where the condition is known to run in families, although this accounts for only around 5% of all cases. An inherited genetic defect on chromosome 21 (coding for superoxide dismutase) is associated with approximately 20% of familial cases of ALS.[6][7] This mutation is believed to be autosomal dominant. The most common ALS causing SOD1 mutation in North America is A4V, characterized by an exceptionally rapid progression from onset to death. The children of those diagnosed with familial ALS have a higher risk factor for developing the disease; however, those who have close family members who have been diagnosed with sporadic ALS have no greater a risk factor than the general population, suggesting again an environmental or other non-genetic cause.[8]
Some environmental causative factors have been suggested for the increased incidence in the western Pacific. Prolonged exposure to a dietary neurotoxin called BMAA is one suspected risk factor in Guam;[9] this neurotoxin produced by cyanobacteria is one of several possible neurotoxic compounds found in the seed of the cycad Cycas circinalis,[10] a tropical plant found in Guam, which was used in the human food supply during the 1950s and early 1960s.
The very high incidence of the disease among Italian soccer players (more than five times higher than normally expected) has raised the concern of a possible link between the disease and the use of pesticides on the soccer fields (several of which have been linked to neuronal toxicity).[11][12] A 2004 Italian study trying to link a high incidence of ALS in soccer players to performance-enhancing drugs failed when the group was compared to cyclists that also used performance-enhancing drugs but without contracting ALS. A possible conclusion was that soccer players experience frequent head trauma (heading the ball, falls and collisions sustained during games) compared to cyclists who wear head protection and rarely have falls.[13][14]
According to the ALS Association, veterans of the United States military are at an increased risk of contracting ALS (again, possibly implying a link to neurotoxic chemical exposure). In its report ALS in the Military,[15] the group pointed to an almost 60% greater chance of the disease in military veterans than the general population. For Gulf War veterans, the chance is seen as twice that of veterans not deployed to the Persian Gulf in a joint study by the Veterans Affairs Administration and the DOD, another epidemiologic association suggesting a link to toxic exposure.[16][17][18]
A 2010 study has raised questions about the diagnosis of ALS in some veterans and athletes, suggesting that repeated concussions may cause a chronic traumatic encephalopathy that mimics ALS; this might explain the higher rate of ALS diagnoses in those populations.[unreliable medical source?][19]
Pathophysiology
Genetic associations include Type OMIM Gene Locus ALS1 105400 SOD1 21q22.1 ALS2 205100 ALS2 2q33.1 ALS3 606640 ? 18q21 ALS4 602433 SETX 9q34.13 ALS5 602099 ? 15q15.1-q21.1 ALS6 608030 FUS 16p11.2 ALS7 608031 ? 20p13 ALS8 608627 VAPB 20q13.3 ALS9 611895 ANG 14q11.2 ALS10 612069 TARDBP 1p36.2 ALS11 612577 FIG4 6q21 ALS12 613435 OPTN 10p15-p14 ALS13 183090 ATXN2 12q24.12 ALS14 613954 VCP 9p13.3 The defining feature of ALS is the death of both upper and lower motor neurons in the motor cortex of the brain, the brain stem, and the spinal cord. Prior to their destruction, motor neurons develop proteinaceous inclusions in their cell bodies and axons. This may be partly due to defects in protein degradation.[20] These inclusions often contain ubiquitin, and generally incorporate one of the ALS-associated proteins: SOD1, TAR DNA binding protein (TDP-43, or TARDBP), or FUS. Interestingly, these inclusions do not stain with the dyes Congo Red or Thioflavin S, and are therefore non-amyloid aggregates.[21][22] This is in contrast to the aggregates and plaques seen in many other neurodegenerative diseases of protein aggregation, including Alzheimer's disease, Parkinson's disease, Huntington's disease, and prion diseases.
SOD1
The cause of ALS is not known, though an important step toward determining the cause came in 1993 when scientists discovered that mutations in the gene that produces the Cu/Zn superoxide dismutase (SOD1) enzyme were associated with some cases (approximately 20%) of familial ALS. This enzyme is a powerful antioxidant that protects the body from damage caused by superoxide, a toxic free radical generated in the mitochondria. Free radicals are highly reactive molecules produced by cells during normal metabolism again largely by the mitochondria. Free radicals can accumulate and cause damage to both mitochondrial and nuclear DNA and proteins within cells. To date, over 110 different mutations in SOD1 have been linked with the disease, some of which have a very long clinical course (e.g. H46R), while others, such as A4V, being exceptionally aggressive. Evidence suggests that failure of defenses against oxidative stress up-regulates programmed cell death (apoptosis), among many other possible consequences. Although it is not yet clear how the SOD1 gene mutation leads to motor neuron degeneration, researchers have theorized that an accumulation of free radicals may result from the faulty functioning of this gene. Current research, however, indicates that motor neuron death is not likely a result of lost or compromised dismutase activity, suggesting mutant SOD1 induces toxicity in some other way (a gain of function).[23][24]
Studies involving transgenic mice have yielded several theories about the role of SOD1 in mutant SOD1 familial amyotrophic lateral sclerosis. Mice lacking the SOD1 gene entirely do not customarily develop ALS, although they do exhibit an acceleration of age-related muscle atrophy (sarcopenia) and a shortened lifespan (see article on superoxide dismutase). This indicates that the toxic properties of the mutant SOD1 are a result of a gain in function rather than a loss of normal function. In addition, aggregation of proteins has been found to be a common pathological feature of both familial and sporadic ALS (see article on proteopathy). Interestingly, in mutant SOD1 mice (most commonly, the G93A mutant), aggregates (misfolded protein accumulations) of mutant SOD1 were found only in diseased tissues, and greater amounts were detected during motor neuron degeneration.[25] It is speculated that aggregate accumulation of mutant SOD1 plays a role in disrupting cellular functions by damaging mitochondria, proteasomes, protein folding chaperones, or other proteins.[26] Any such disruption, if proven, would lend significant credibility to the theory that aggregates are involved in mutant SOD1 toxicity. Critics have noted that in humans, SOD1 mutations cause only 2% or so of overall cases and the etiological mechanisms may be distinct from those responsible for the sporadic form of the disease. To date, the ALS-SOD1 mice remain the best model of the disease for preclinical studies but it is hoped that more useful models will be developed.
Lactate Dyscrasia Hypothesis
Researchers specializing in the neurobiology of aging have proposed a novel molecular model for the pathogenesis of ALS called the lactate dyscrasia hypothesis that involves an adenosine triphosphate (ATP)-dependent muscle neuronal lactate shuttle (MNLS) at the neuromuscular junction (NMJ) to regulate the flow of lactate from muscle to neurons and vice versa. Failure of the MNLS due to respiratory chain dysfunction is proposed to result in lactate toxicity and degeneration of nerve endings at the NMJ leading to nerve terminus dysjunction from the muscle cell.[27]
Other factors
Studies also have focused on the role of glutamate in motor neuron degeneration. Glutamate is one of the chemical messengers or neurotransmitters in the brain. Scientists have found that, compared to healthy people, ALS patients have higher levels of glutamate in the serum and spinal fluid.[7] Riluzole is currently the only FDA approved drug for ALS and targets glutamate transporters. It only has a modest effect on survival, however, suggesting that excess glutamate is not the sole cause of the disease.
Diagnosis
No test can provide a definite diagnosis of ALS, although the presence of upper and lower motor neuron signs in a single limb is strongly suggestive. Instead, the diagnosis of ALS is primarily based on the symptoms and signs the physician observes in the patient and a series of tests to rule out other diseases. Physicians obtain the patient's full medical history and usually conduct a neurologic examination at regular intervals to assess whether symptoms such as muscle weakness, atrophy of muscles, hyperreflexia, and spasticity are getting progressively worse.
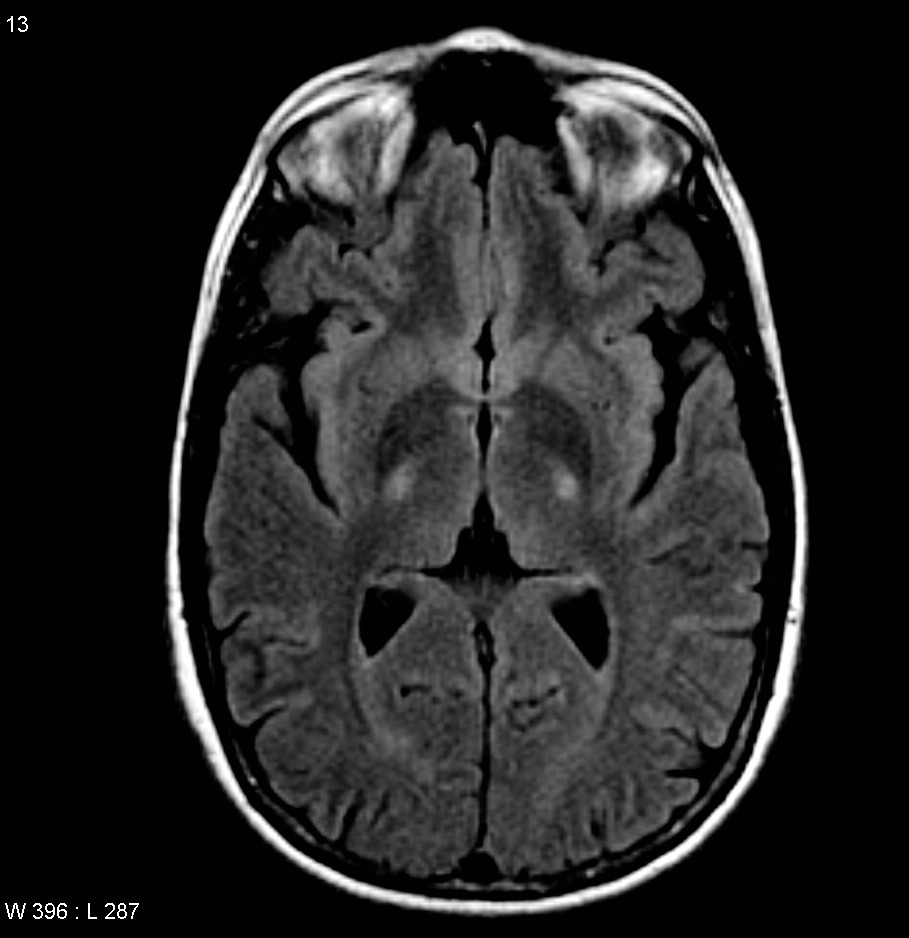
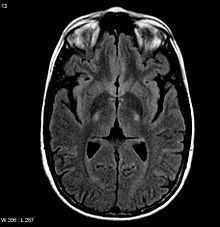 MRI (axial FLAIR) demonstrates increased T2 signal within the posterior part of the internal capsule, consistent with the clinical diagnosis of ALS.
MRI (axial FLAIR) demonstrates increased T2 signal within the posterior part of the internal capsule, consistent with the clinical diagnosis of ALS.
Because symptoms of ALS can be similar to those of a wide variety of other, more treatable diseases or disorders, appropriate tests must be conducted to exclude the possibility of other conditions. One of these tests is electromyography (EMG), a special recording technique that detects electrical activity in muscles. Certain EMG findings can support the diagnosis of ALS. Another common test measures nerve conduction velocity (NCV). Specific abnormalities in the NCV results may suggest, for example, that the patient has a form of peripheral neuropathy (damage to peripheral nerves) or myopathy (muscle disease) rather than ALS. The physician may order magnetic resonance imaging (MRI), a noninvasive procedure that uses a magnetic field and radio waves to take detailed images of the brain and spinal cord. Although these MRI scans are often normal in patients with ALS, they can reveal evidence of other problems that may be causing the symptoms, such as a spinal cord tumor, multiple sclerosis, a herniated disk in the neck, syringomyelia, or cervical spondylosis.
Based on the patient's symptoms and findings from the examination and from these tests, the physician may order tests on blood and urine samples to eliminate the possibility of other diseases as well as routine laboratory tests. In some cases, for example, if a physician suspects that the patient may have a myopathy rather than ALS, a muscle biopsy may be performed.
Infectious diseases such as human immunodeficiency virus (HIV), human T-cell leukaemia virus (HTLV), Lyme disease,[28] syphilis[29] and tick-borne encephalitis[30] viruses can in some cases cause ALS-like symptoms. Neurological disorders such as multiple sclerosis, post-polio syndrome, multifocal motor neuropathy, CIDP, and spinal muscular atrophy can also mimic certain facets of the disease and should be considered by physicians attempting to make a diagnosis.
Because of the prognosis carried by this diagnosis and the variety of diseases or disorders that can resemble ALS in the early stages of the disease, patients should always obtain a second neurological opinion.
Treatment
Slowing progression
Riluzole (Rilutek) as of 2011 is the only treatment that has been found to improve survival but only to a modest extent.[31] It lengthens survival by several months, and may have a greater survival benefit for those with a bulbar onset. It also extends the time before a person needs ventilation support. Riluzole does not reverse the damage already done to motor neurons, and people taking it must be monitored for liver damage (occurring in ~10% of people taking the drug).[32] It is approved by Food and Drug Administration (FDA) and recommended by the National Institute for Clinical Excellence (NICE).
Symptomatic
Other treatments for ALS are designed to relieve symptoms and improve the quality of life for patients. This supportive care is best provided by multidisciplinary teams of health care professionals such as physicians; pharmacists; physical, occupational, and speech therapists; nutritionists; social workers; and home care and hospice nurses. Working with patients and caregivers, these teams can design an individualized plan of medical and physical therapy and provide special equipment aimed at keeping patients as mobile and comfortable as possible.
Medical professionals can prescribe medications to help reduce fatigue, ease muscle cramps, control spasticity, and reduce excess saliva and phlegm. Drugs also are available to help patients with pain, depression, sleep disturbances, dysphagia, and constipation.
Physical therapists and occupational therapists play a large role in rehabilitation for individuals with ALS. Specifically, physical and occupational therapists can set goals and promote benefits for individuals with ALS by delaying loss of strength, maintaining endurance, limiting pain, preventing complications, and promoting functional independence.[33] There is also a strong emphasis on the importance of patient and caregiver education that can be reinforced by physical therapists or occupational therapists.[33] Research is controversial as to whether implementing a specific exercise program for these individuals may be beneficial; moreover, it is important for a physical therapist to address and understand the risks associated with implementing these types of programs for each and every person with ALS and the severity of their condition. The controversy lies in the fact that because ALS is characteristic of the degeneration of upper and lower motor neurons, that these neurons may react differently to specific exercise programs.[33] Because spasticity is a common characteristic for individuals with ALS, physical therapists aim to reduce this by implementing range of motion activities with minimal resistance.[33] In addition to range of motion activities, positioning techniques and splinting have also been shown to reduce spasticity; moreover, these techniques can also play an integral role in the reduction of pain for people with ALS.[33] Overall, physical therapists have been proven to have positive effects on individuals with ALS by prescribing techniques and equipment to assist with conserving energy, emphasizing the importance of education, limiting pain, and help to maintain a level of function appropriate for each of their clients with ALS.[33]
Occupational therapy and special equipment such as assistive technology can also enhance patients' independence and safety throughout the course of ALS. But physical therapists must be mindful when prescribing assistive devices, keeping in mind the patients and their attitudes. Devices should make the patient feel hopeful, not helpless. Gentle, low-impact aerobic exercise such as walking, swimming, and stationary bicycling can strengthen unaffected muscles, improve cardiovascular health, and help patients fight fatigue and depression. Range of motion and stretching exercises can help prevent painful spasticity and shortening (contracture) of muscles. Physical therapists can recommend exercises that provide these benefits without overworking muscles. They can suggest devices such as ramps, braces, walkers, and wheelchairs that help patients remain mobile. Examples of devices prescribed can include cervical collars.[34] In ALS, there will be a progression of cervical extensor weakness.[34] Weakness of the muscles will cause the patient's head to fall forward, leading to acute neck pain, potential for chronic cervical conditions to develop and tightness of anterior neck muscles.[34] A forward head posture will interfere in patients ADLs, making them more dependent on caretakers. A cervical collar can help restore their independence and comfort. When there is mild to moderate weakness of the cervical extensor, the therapist may provide a soft foam collar.[34] When more severe weakness is observed, a more rigid collar will be beneficial.[34] Occupational therapists can provide or recommend equipment and adaptations to enable people to retain as much independence in activities of daily living as possible.
ALS patients who have difficulty speaking may benefit from working with a speech-language pathologist. These health professionals can teach patients adaptive strategies such as techniques to help them speak louder and more clearly. As ALS progresses, speech-language pathologists can recommend the use of augmentative and alternative communication such as voice amplifiers, speech-generating devices (or voice output communication devices) and/or low tech communication techniques such as alphabet boards or yes/no signals. These methods and devices help patients communicate when they can no longer speak or produce vocal sounds. With the help of occupational therapists, speech-generating devices can be activated by switches or mouse emulation techniques controlled by small physical movements of, for example, the head, finger or eyes. In every case, the appropriate therapist should be mindful of the patients' preferences, attitudes, and likely progression over time.
Patients and caregivers can learn from speech-language pathologists and nutritionists how to plan and prepare numerous small meals throughout the day that provide enough calories, fiber, and fluid and how to avoid foods that are difficult to swallow. Patients may begin using suction devices to remove excess fluids or saliva and prevent choking. When patients can no longer get enough nourishment from eating, doctors may advise inserting a feeding tube into the stomach. The use of a feeding tube also reduces the risk of choking and pneumonia that can result from inhaling liquids into the lungs. The tube is not painful and does not prevent patients from eating food orally if they wish.
When the muscles that assist in breathing weaken, use of ventilatory assistance (intermittent positive pressure ventilation (IPPV), bilevel positive airway pressure (BIPAP), or biphasic cuirass ventilation (BCV)) may be used to aid breathing. Such devices artificially inflate the patient's lungs from various external sources that are applied directly to the face or body. When muscles are no longer able to maintain oxygen and carbon dioxide levels, these devices may be used full-time. BCV has the added advantage of being able to assist in clearing secretions by using high-frequency oscillations followed by several positive expiratory breaths.[35] Patients may eventually consider forms of mechanical ventilation (respirators) in which a machine inflates and deflates the lungs. To be effective, this may require a tube that passes from the nose or mouth to the windpipe (trachea) and for long-term use, an operation such as a tracheostomy, in which a plastic breathing tube is inserted directly in the patient's windpipe through an opening in the neck.
Patients and their families should consider several factors when deciding whether and when to use one of these options. Ventilation devices differ in their effect on the patient's quality of life and in cost. Although ventilation support can ease problems with breathing and prolong survival, it does not affect the progression of ALS. Patients need to be fully informed about these considerations and the long-term effects of life without movement before they make decisions about ventilation support. Some patients under long-term tracheostomy intermittent positive pressure ventilation with deflated cuffs or cuffless tracheostomy tubes (leak ventilation) are able to speak, provided their bulbar muscles are strong enough. This technique preserves speech in some patients with long-term mechanical ventilation.
Social workers and home care and hospice nurses help patients, families, and caregivers with the medical, emotional, and financial challenges of coping with ALS, particularly during the final stages of the disease. Social workers provide support such as assistance in obtaining financial aid, arranging durable power of attorney, preparing a living will, and finding support groups for patients and caregivers. Home nurses are available not only to provide medical care but also to teach caregivers about tasks such as maintaining respirators, giving feedings, and moving patients to avoid painful skin problems and contractures. Home hospice nurses work in consultation with physicians to ensure proper medication, pain control, and other care affecting the quality of life of patients who wish to remain at home. The home hospice team can also counsel patients and caregivers about end-of-life issues.
Researchers have stated that "ALS patients have a chronically deficient intake of energy and recommended augmentation of energy intake."[36] Both animal [37] and human research [36][38] suggest that ALS patients should be encouraged to consume as many calories as possible and not to restrict their calorie intake.
Many ALS patients use complementary and alternative medicines in an attempt to slow their disease.[citation needed] This may include popular vitamin supplements such as Vitamin C, high doses of vitamins and nutrients ("mega-dosing"), traditional Chinese medicine, or other forms of therapy such as acupuncture, reiki, or massage. To date there have been no studies demonstrating that such treatment approaches have an effect on the progression of the disease. Given the lack of therapeutic options, people with ALS can be vulnerable to snake oil scams involving complicated medical terminology or potentially exciting technologies such as stem cell transplantation. Practitioners of these scams promise amazing results but carry out little or no real follow up or study of the patients they have treated in order to prove their assertions. The risks of false hope, financial harm, and potentially medically harm, are a threat to the wellbeing of ALS patients and their families.
Prognosis
Eventually most people with ALS are not able to stand or walk, get in or out of bed on their own, use their hands and arms, or communicate. In later stages of the disease, individuals have difficulty breathing as the muscles of the respiratory system weaken. Although respiratory support can ease problems with breathing and prolong survival, it does not affect the progression of ALS. Most people with ALS die from respiratory failure, usually within three to five years from the onset of symptoms. The median survival time from onset to death ranges from 20 to 48 months, but 10 to 20% of ALS patients have a survival longer than 10 years.[39] The world's most widely recognized person with ALS, Stephen Hawking, has lived with the disease for more than 40 years, though he is an unusual case.[40]
Epidemiology
ALS is one of the most common neuromuscular diseases worldwide, and people of all races and ethnic backgrounds are affected. One or two out of 100,000 people develop ALS each year.[41] ALS most commonly strikes people between 40 and 60 years of age, but younger and older people can also develop the disease. Men are affected slightly more often than women.
"Familial ALS" accounts for approximately 5%–10% of all ALS cases and is caused by genetic factors. Of these, approximately 1 in 10 is linked to a mutation in copper/zinc superoxide dismutase (SOD1), an enzyme responsible for scavenging free radicals. A recent study has identified a gene called FUS ("Fused in Sarcoma", ALS6) as being responsible for 1 in 20 cases of fALS.[42][43]
Although the incidence of ALS is thought to be regionally uniform, there are three regions in the West Pacific where there has in the past been an elevated occurrence of ALS. This seems to be declining in recent decades. The largest is the area of Guam inhabited by the Chamorro people, who have historically had a high incidence (as much as 143 cases per 100,000 people per year) of a condition called Lytico-Bodig disease which is a combination of ALS, Parkinsonism, and dementia.[44] Two more areas of increased incidence are West Papua and the Kii Peninsula of Japan.[45][46]
Although there have been reports of several "clusters" including three American football players from the San Francisco 49ers, more than fifty football players in Italy,[11] three football-playing friends in the south of England,[47] and reports of conjugal (husband and wife) cases in the south of France,[48][49][50][51][52] these are statistically plausible chance events[citation needed]. Although many authors consider ALS to be caused by a combination of genetic and environmental risk factors, so far the latter have not been firmly identified, other than a higher risk with increasing age.
In 2010, a VA study found that head trauma can produce symptoms that resemble ALS but that are actually chronic traumatic encephalopathy (CTE). Postmortem brain studies conducted on two American football players showed evidence of CTE, rather than ALS.[53]
Etymology
Amyotrophic comes from the Greek language: A- means "no", myo refers to "muscle", and trophic means "nourishment"; amyotrophic therefore means "no muscle nourishment," which describes the characteristic atrophication of the sufferer's disused muscle tissue. Lateral identifies the areas in a person's spinal cord where portions of the nerve cells that are affected are located. As this area degenerates it leads to scarring or hardening ("sclerosis") in the region.
History
Timeline Year Event 1850 English scientist Augustus Waller describes the appearance of shriveled nerve fibers 1869 French doctor Jean-Martin Charcot first[dubious ] describes ALS in scientific literature[54] 1881 "On Amyotrophic Lateral Sclerosis" is translated into English and published in a three-volume edition of Lectures on the Diseases of the Nervous System 1939 ALS becomes a cause célèbre in the United States when baseball legend Lou Gehrig's career—and, two years later, his life—is ended by the disease. He gives his farewell speech on July 4. 1950s ALS epidemic occurs among the Chamorro people on Guam 1991 Researchers link chromosome 21 to FALS (Familial ALS) 1993 SOD1 gene on chromosome 21 found to play a role in some cases of FALS 1996 Rilutek becomes the first FDA-approved drug for ALS 1998 The El Escorial criteria is developed as the standard for classifying ALS patient in clinical research Clinical Research
A number of clinical trials are underway globally for ALS; a comprehensive listing of trials in the US can be found at ClinicalTrials.gov.
KNS-760704 is under clinical investigation in ALS patients. It is hoped that the drug will have a neuroprotective effect. It is one enantiomer of pramipexole, which is approved for the treatment of Parkinson's disease and restless legs syndrome.[55] The single-enantiomer preparation is essentially inactive at dopamine receptors, is not dose limited by the potent dopaminergic properties of pramipexole.[56] Results of a Phase II clinical trial conducted by Knopp Neurosciences and involving 102 patients were reported in 2010; the trial found a dose-dependent slowing in loss of function.[57]
Olesoxime (TRO19622) is being tested in a phase 3 clinical study by Trophos, French biotechnology company, as part of the MitoTarget Project.[58] The molecule has a cholesterol-like structure and displays strong neuroprotective properties, and it should be as effective as a cocktail of three neurotrophic factors in keeping motor neurons alive in culture. The ongoing clinical study aims to test efficacy, safety, tolerability and plasma levels in patients with ALS, to see whether a single daily dose of two capsules – under certain circumstances – can improve survival and symptoms of ALS patients. The trial started in May 2009, all the patients are recruited and results are expected in the last quarter of 2011. The study is taking place in France, Belgium, Germany, the UK and Spain.
Talampanel is being tested in ALS by Teva Pharmaceutical Industries; a Phase II trial was completed in April 2010. [59]
The new discovery of RNAi has some promise in treating ALS. In recent studies, RNAi has been used in lab rats to shut off specific genes that lead to ALS. Cytrx Corporation has sponsored ALS research using RNAi gene silencing technology targeted at the mutant SOD1 gene.[60] Cytrx's orally-administered drug Arimoclomol is currently in clinical evaluation as a therapeutic treatment for ALS.
See also
- ALS Association
- ALS Society of Canada
- ALS Therapy Development Institute
- Multifocal motor neuropathy, a condition often mistaken for ALS
- Muscular Dystrophy Association
- The Aging Brain
References
- ^ "amyotrophic lateral sclerosis" at Dorland's Medical Dictionary
- ^ Rowland, L.P. (1994). "Amyotrophic lateral sclerosis: theories and therapies". Ann. Neurol. 35 (2): 129–130. doi:10.1002/ana.410350202. PMID 8109893.
- ^ Phukan J, Pender NP, Hardiman O (2007). "Cognitive impairment in amyotrophic lateral sclerosis". Lancet Neurol 6 (11): 994–1003. doi:10.1016/S1474-4422(07)70265-X. PMID 17945153. http://linkinghub.elsevier.com/retrieve/pii/S1474-4422(07)70265-X.
- ^ www.alsa.org
- ^ Mateen FJ, Carone M, Sorenson EJ (October 2010). "Patients who survive 5 years or more with ALS in Olmsted County, 1925-2004.". J Neurol Neurosurg Psychiatry 81 (10): 1144–6. doi:10.1136/jnnp.2009.201251. PMC 2946435. PMID 20627966. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2946435.
- ^ Conwit, Robin A. (December 2006). "Preventing familial ALS: A clinical trial may be feasible but is an efficacy trial warranted?". Journal of the Neurological Sciences 251 (1–2): 1–2. doi:10.1016/j.jns.2006.07.009. ISSN 0022-510X. PMID 17070848.
- ^ a b Al-Chalabi, Ammar; P. Nigel Leigh (August 2000). "Recent advances in amyotrophic lateral sclerosis". Current Opinion in Neurology 13 (4): 397–405. doi:10.1097/00019052-200008000-00006. ISSN 1473-6551. PMID 10970056.
- ^ "ALS Clinic at the Penn State Milton S. Hershey Medical Center". Web.archive.org. Archived from the original on 2004-11-15. http://web.archive.org/web/20041115214832/http://www.alsphiladelphia.org/pennstatehershey/newsletters/newsletter_spring04.htm. Retrieved 2010-06-13.
- ^ "Cyanobacterial neurotoxin BMAA in ALS and Alzheimer's disease". http://www.ncbi.nlm.nih.gov/pubmed/19254284.
- ^ Khabazian I, Bains JS, Williams DE, Cheung J, Wilson JM, Pasqualotto BA, Pelech SL, Andersen RJ, Wang YT, Liu L, Nagai A, Kim SU, Craig UK, Shaw CA (August 2002). "Isolation of various forms of sterol beta-D-glucoside from the seed of Cycas circinalis: neurotoxicity and implications for ALS-parkinsonism dementia complex". J. Neurochem. 82 (3): 516–28. doi:10.1046/j.1471-4159.2002.00976.x. PMID 12153476. http://www3.interscience.wiley.com/resolve/openurl?genre=article&sid=nlm:pubmed&issn=0022-3042&date=2002&volume=82&issue=3&spage=516. Retrieved 2009-03-13.
- ^ a b "Sla, indagini nei club. Pesticidi nel mirino". http://www.corriere.it/sport/08_ottobre_03/sla_indagine_pesticidi_fd04f986-911c-11dd-9f28-00144f02aabc.shtml. Retrieved 2008-10-02.
- ^ "Sla, una strage nel calcio". http://www.gazzetta.it/Calcio/Altro_Calcio/Primo_Piano/2007/11_Novembre/30/sla_3011.shtml. Retrieved 2008-10-02.
- ^ http://www.nwitimes.com/sports/columnists/john-doherty/article_4cae14e5-4ae0-5c76-b717-af761f19bc24.html JOHN DOHERTY: ALS link to concussions not new
- ^ "Soccer, neurotrauma and amyotrophic lateral sclerosis: is there a connection?". http://www.ncbi.nlm.nih.gov/pubmed/15119987.
- ^ "ALS in the Military". The ALS Association. 2007-05-17. http://www.alsa.org/files/pdf/als_military_paper.pdf. Retrieved 2008-05-01.
- ^ "Occurrence of ALS higher in Gulf War veterans". http://www.bcm.edu/fromthelab/vol02/is10/03oct_n1.htm.
- ^ "Veterans get ALS disability". http://www.baltimoresun.com/news/nation/bal-te.als25jul25,0,7448073.story.
- ^ "ALS Research: Poison Dirt?". http://quest.mda.org/news/als-research-poison-dirt.
- ^ McKee AC, Gavett BE, Stern RA, Nowinski CJ, Cantu RC, Kowall NW, Perl DP, Hedley-Whyte ET, Price B, Sullivan C, Morin P, Lee HS, Kubilus CA, Daneshvar DH, Wulff M, Budson AE. (August 2010). "TDP-43 Proteinopathy and Motor Neuron Disease in Chronic Traumatic Encephalopathy.". J Neuropathol Exp Neurol.. http://journals.lww.com/jneuropath/Abstract/publishahead/TDP_43_Proteinopathy_and_Motor_Neuron_Disease_in.99709.aspx. Retrieved 2010-08-23.
- ^ Deng, HX; Chen, W, Hong, ST, Boycott, KM, Gorrie, GH, Siddique, N, Yang, Y, Fecto, F, Shi, Y, Zhai, H, Jiang, H, Hirano, M, Rampersaud, E, Jansen, GH, Donkervoort, S, Bigio, EH, Brooks, BR, Ajroud, K, Sufit, RL, Haines, JL, Mugnaini, E, Pericak-Vance, MA, Siddique, T (2011-08-21). "Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia.". Nature 477 (7363). doi:10.1038/nature10353. PMID 21857683.
- ^ Kwong LK, Uryu K, Trojanowski JQ, Lee VM (2008). "TDP-43 proteinopathies: neurodegenerative protein misfolding diseases without amyloidosis". Neurosignals 16 (1): 41–51. doi:10.1159/000109758. PMID 18097159.
- ^ Kerman A, Liu HN, Croul S, Bilbao J, Rogaeva E, Zinman L, Robertson J, Chakrabartty A (2010). "Amyotrophic lateral sclerosis is a non-amyloid disease in which extensive misfolding of SOD1 is unique to the familial form". Acta Neuropathol. 119 (3): 335–44. doi:10.1007/s00401-010-0646-5. PMID 20111867.
- ^ Reaume A, Elliott J, Hoffman E, Kowall N, Ferrante R, Siwek D, Wilcox H, Flood D, Beal M, Brown R, Scott R, Snider W (1996). "Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury". Nat Genet 13 (1): 43–7. doi:10.1038/ng0596-43. PMID 8673102.
- ^ Bruijn L, Houseweart M, Kato S, Anderson K, Anderson S, Ohama E, Reaume A, Scott R, Cleveland D (1998). "Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1". Science 281 (5384): 1851–4. doi:10.1126/science.281.5384.1851. PMID 9743498.
- ^ Furukawa Y, Fu R, Deng H, Siddique T, O'Halloran T (2006). "Disulfide cross-linked protein represents a significant fraction of ALS-associated Cu, Zn-superoxide dismutase aggregates in spinal cords of model mice". Proc Natl Acad Sci USA 103 (18): 7148–53. doi:10.1073/pnas.0602048103. PMC 1447524. PMID 16636274. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1447524.
- ^ Boillée S, Vande Velde C, Cleveland D (2006). "ALS: a disease of motor neurons and their nonneuronal neighbors". Neuron 52 (1): 39–59. doi:10.1016/j.neuron.2006.09.018. PMID 17015226.
- ^ Meethal, SV, Atwood, CS (April 2010). "Lactate dyscrasia: a novel explanation for amyotrophic lateral sclerosis". Neurobiology of Aging 20. doi:10.1016/j.neurobiolaging.2010.04.012.
- ^ Hansel Y, Ackerl M, Stanek G. (1995). "ALS-like sequelae in chronic neuroborreliosis". Wien Med Wochenschr. 145 (7-8): 186–8. PMID 7610670.
- ^ el Alaoui-Faris M, Medejel A, al Zemmouri K, Yahyaoui M, Chkili T (1990). "Amyotrophic lateral sclerosis syndrome of syphilitic origin. 5 cases". Rev Neurol (Paris) 146 (1): 41–4. PMID 2408129.
- ^ Umanekii KG, Dekonenko EP (1983). "Structure of progressive forms of tick-borne encephalitis". Zh Nevropatol Psikhiatr Im S S Korsakova. 83 (8): 1173–9. PMID 6414202.
- ^ Carlesi, C; Pasquali, L, Piazza, S, Lo Gerfo, A, Caldarazzo Ienco, E, Alessi, R, Fornai, F, Siciliano, G (2011 Mar). "Strategies for clinical approach to neurodegeneration in Amyotrophic lateral sclerosis.". Archives italiennes de biologie 149 (1): 151-67. PMID 21412722.
- ^ Miller, RG; Mitchell JD, Lyon M, Moore DH, G (2007). "Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND).". Cochrane Database of Systematic Reviews (1). PMID 17253460.
- ^ a b c d e f Lewis, M. & Rushanan, S. (2007). "The role of physical therapy and occupational therapy in the treatment of Amyotrophic Lateral Sclerosis". NeuroRehabilitation 22: 451–461.
- ^ a b c d e O'Sullivan, Susan B.; Thomas J. Schmitz (2007). Physical Rehabilitation (Fifth Edition ed.). Philadelphia: Davis Company. pp. 835–836.
- ^ Sviri S, Linton DM, Van Heerden PV (Jun 2005). "Non-invasive Mechanical Ventilation Enhances Patient Autonomy in Decision-Making Regarding Chronic Ventilation". Critical Care and Resuscitation 7 (2): 116–118. PMID 16548804.
- ^ a b Kasarskis EJ, Berryman S, Vanderleest JG, Schneider AR, McClain CJ (Jan 1996). "Nutritional status of patients with amyotrophic lateral sclerosis: relation to the proximity of death". Am J Clin Nutr. 63 (1): 130–7. PMID 8604660. http://www.ajcn.org/cgi/pmidlookup?view=long&pmid=8604660.
- ^ Hamadeh MJ, Rodriguez MC, Kaczor JJ, Tarnopolsky MA (Feb 2005). "Caloric restriction transiently improves motor performance but hastens clinical onset of disease in the Cu/Zn-superoxide dismutase mutant G93A mouse". Muscle Nerve 31 (2): 214–20. doi:10.1002/mus.20255. PMID 15625688.
- ^ Slowie LA, Paige MS, Antel JP (Jul 1983). "Nutritional considerations in the management of patients with amyotrophic lateral sclerosis (ALS)". J Am Diet Assoc 83 (1): 44–7. PMID 6863783.
- ^ Prognostic factors in ALS: A critical review published in Amyotrophic Lateral Sclerosis Journal, 05 December 2008, Adriano Chio et al.
- ^ http://articles.cnn.com/2009-04-20/health/hawking.als_1_als-association-lou-gehrig-s-disease-motor-neuron-disease?_s=PM:HEALTH
- ^ "ALS Topic Overview". http://www.webmd.com/brain/tc/Amyotrophic-Lateral-Sclerosis-ALS-Topic-Overview. Retrieved 2008-05-01.
- ^ Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, Shaw CE (February 2009). "Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6". Science 323 (5918): 1208–11. doi:10.1126/science.1165942. PMID 19251628. http://www.sciencemag.org/cgi/pmidlookup?view=long&pmid=19251628. Retrieved 2009-03-13.
- ^ Kwiatkowski TJ, Bosco DA, LeClerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown, RH (Feb 2009). "Mutations in the FUS/TLS Gene on Chromosome 16 Cause Familial Amyotrophic Lateral Sclerosis". Science 323 (5918): 1205–1208. doi:10.1126/science.1166066. PMID 19251627.
- ^ Reed D, Labarthe D, Chen KM, Stallones R (Jan 1987). "A cohort study of amyotrophic lateral sclerosis and parkinsonism-dementia on Guam and Rota". Am J Epidemiol. 125 (1): 92–100. PMID 3788958. http://aje.oxfordjournals.org/cgi/pmidlookup?view=long&pmid=3788958.
- ^ S. Kuzuhara, Y. Kokubo P3-146Marked increase of parkinsonism-dementia (P-D) phenotypes in the high incidence amyotrophic lateral sclerosis (ALS) focus in the Kii peninsula of Japan. Alzheimer's and Dementia, Volume 2, Issue 3, Pages S417-S417
- ^ Spencer PS, Palmer VS, Ludolph AC (Aug 2005). "On the decline and etiology of high-incidence motor system disease in West Papua (southwest New Guinea)". Mov. Disord. 20 (Suppl 12): S119–26. doi:10.1002/mds.20552. PMID 16092101.
- ^ Wicks P, Abrahams S, Masi D, Hejda-Forde S, Leigh PN & Goldstein LH (2005) The Prevalence of Depression and Anxiety in MND, Amyotrophic Lateral Sclerosis and other Motor Neuron Disorders, Volume 6, Supplement 1, p. 147
- ^ Rachele MG, Mascia V, Tacconi P, Dessi N, Marrosu F (April 1998). "Conjugal amyotrophic lateral sclerosis: a report on a couple from Sardinia, Italy". Ital J Neurol Sci. 19 (2): 97–100. doi:10.1007/BF02427565. PMID 10935845.
- ^ Poloni M, Micheli A, Facchetti D, Mai R, Ceriani F (April 1997). "Conjugal amyotrophic lateral sclerosis: toxic clustering or change?". Ital J Neurol Sci. 18 (2): 109–12. doi:10.1007/BF01999572. PMID 9239532.
- ^ Camu W, Cadilhac J, Billiard M. (March 1994). "Conjugal amyotrophic lateral sclerosis: a report on two couples from southern France". Neurology 44 (3 Pt 1): 547–8. PMID 8145930.
- ^ Cornblath DR, Kurland LT, Boylan KB, Morrison L, Radhakrishnan K, Montgomery M. (November 1993). "Conjugal amyotrophic lateral sclerosis: report of a young married couple". Neurology 43 (11): 2378–80. PMID 8232960.
- ^ Corcia P, Jafari-Schluep HF, Lardillier D, Mazyad H, Giraud P, Clavelou P, Pouget J, Camu W (November 2003). "A clustering of conjugal amyotrophic lateral sclerosis in southeastern France". Neurol. 60 (4): 553–7. doi:10.1001/archneur.60.4.553. PMID 12707069.
- ^ Schwarz, Alan. "Study Says Brain Trauma Can Mimic A.L.S.", The New York Times, August 18, 2010. Retrieved August 18, 2010.
- ^ Rowland LP (March 2001). "How amyotrophic lateral sclerosis got its name: the clinical-pathologic genius of Jean-Martin Charcot". Arch. Neurol. 58 (3): 512–5. doi:10.1001/archneur.58.3.512. PMID 11255459. http://archneur.ama-assn.org/cgi/content/extract/58/3/512.
- ^ Abramova NA et al. Inhibition by R(+) or S(-) pramipexole of caspase activation and cell death induced by methylpyridinium ion or beta amyloid peptide in SH-SY5Y neuroblastoma. J Neurosci Res. 2002 Feb 15;67(4):494-500.
- ^ Gribkoff V and Bozik M. KNS-760704 [(6R)-4,5,6,7-tetrahydro-N6-propyl-2, 6-benzothiazole-diamine dihydrochloride monohydrate] for the treatment of amyotrophic lateral sclerosis. CNS Neurosci Ther. 2008 Fall;14(3):215-26.
- ^ http://journals.lww.com/neurotodayonline/Fulltext/2010/07010/New_Als_Drug_Shows_Dose_Dependent_Efficacy_in.2.aspx
- ^ http://ec.europa.eu/research/health/medical-research/brain-research/projects/mitotarget_en.html
- ^ http://clinicaltrials.gov/ct2/show/NCT00696332?term=ALS&rank=2
- ^ Xia X, Zhou H, Huang Y, Xu Z (Sep 2006). "Allele-specific RNAi selectively silences mutant SOD1 and achieves significant therapeutic benefit in vivo". Neurobiol Dis. 23 (3): 578–86. doi:10.1016/j.nbd.2006.04.019. PMID 16857362.
Further reading
- "ALS Hope Foundation". http://www.alshopefoundation.org/. Retrieved 2008-06-21. "Dedicated to the care and cure of people with Lou Gehrig's Disease. (from site page us.php)"
- "Lou Gehrig: The Official Web Site". CMG Worldwide. http://www.lougehrig.com. Retrieved 2008-06-21. "The Official Web site of Lou Gehrig is an informational Web site intended to honor the life, the legend and the career of Lou Gehrig. (from site page siteinfo/index.htm)"
- Patrick Aebischer; Ann C. Kato (November 2007). "Playing defense against Lou Gehrig's Disease" (Paper). Scientific American (Verlagsgruppe Georg von Holtzbrinck): pp. 86–93. http://www.sciam.com/article.cfm?id=playing-defense-against-l. Retrieved 2008-06-21. "Researchers have proposed potential therapies for a paralyzing disorder once thought to be untreatable (sub-title)"
External links
Categories:- Motor neurone disease
- Rare diseases
- Unsolved problems in neuroscience
- Systemic atrophies primarily affecting the central nervous system
- Cytoskeletal defects
Wikimedia Foundation. 2010.