- Metachromatic leukodystrophy
-
Metachromatic leukodystrophy Classification and external resources 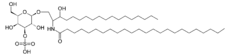
SulfatideICD-10 E75.2 ICD-9 330.0 OMIM 250100 DiseasesDB 8080 eMedicine ped/2893 MeSH D007966 Metachromatic leukodystrophy (MLD, also called Arylsulfatase A deficiency) is a lysosomal storage disease which is commonly listed in the family of leukodystrophies. Leukodystrophies affect the growth and/or development of myelin, the fatty covering which acts as an insulator around nerve fibers throughout the central and peripheral nervous systems. It involves sulfatide accumulation.[1]
Contents
Causes
MLD is directly caused by a deficiency of the enzyme arylsulfatase A[2] and is usually characterized by enzyme activity which is less than 10% of human controls.[3] Without this enzyme, sulfatides build up in many tissues of the body, eventually destroying the myelin sheath of the nervous system. The myelin sheath is a fatty covering that protects nerve fibers. Without it, the nerves in the brain (central nervous system - CNS) and the peripheral nerves (peripheral nervous system - PNS) which control, among other things the muscles related to mobility, cease to function properly.
A recent study contended sulfatide is not completely responsible for MLD because it is non toxic. It has been suggested lysosulfatide, sulfatide which has had its acyl group removed, plays a role because of its cytotoxic properties in vitro.[4]
Genetics


MLD has an autosomal recessive inheritance pattern. The inheritance probabilities per birth are as follows:
- If both parents are carriers:
- 25% (1 in 4) children will have the disorder
- 50% (2 in 4) children will be carriers (but unaffected)
- 25% (1 in 4) children will be free of MLD - unaffected child that is not a carrier
- If one parent is affected and one is free of MLD:
- 0% (0) children will have the disorder - only one parent is affected, other parent always gives normal gene
- 100% (4 in 4) children will be carriers (but unaffected)
- If one parent is a carrier and the other is free of MLD:
- 50% (2 in 4) children will be carriers (but unaffected)
- 50% (2 in 4) children will be free of MLD - unaffected child that is not a carrier
In addition to these frequencies there is a 'pseudo'-deficiency that affects 7% of the population. People with the pseudo deficiency do not have any MLD problems unless they also have carrier or affected status. Pseudo-deficiency tests as low enzyme levels but sulfatide is processed normally so MLD symptoms do not exist.
For further information, see recessive gene and dominance relationship. Also, consult the MLD genetics page at the MLD Foundation.
Symptoms and forms
Like many other genetic disorders that affect lipid metabolism, there are several forms of MLD, which are late infantile, juvenile, and adult.
- In the late infantile form, which is the most common form of MLD (50-60%), affected children begin having difficulty walking after the first year of life, usually at 15–24 months. Symptoms include muscle wasting and weakness, muscle rigidity, developmental delays, progressive loss of vision leading to blindness, convulsions, impaired swallowing, paralysis, and dementia. Children may become comatose. Untreated, most children with this form of MLD die by age 5, often much sooner.
- Children with the juvenile form of MLD (onset between 3–10 years of age) usually begin with impaired school performance, mental deterioration, and dementia and then develop symptoms similar to the late infantile form but with slower progression. Age of death is variable, but normally within 10 to 15 years of symptom onset although some juveniles can live for several decades or longer after onset.
- The adult form commonly begins after age 16 as a psychiatric disorder or progressive dementia. Adult-onset MLD progresses more slowly than the late infantile and juvenile forms, with a protracted course of a decade or more.
Palliative care can help with many of the symptoms and usually improves quality and longevity of life.
Carriers have low enzyme levels compared to their family population ("normal" levels vary from family to family) but even low enzyme levels are adequate to process the body's sulfatide.
Treatment
There is no cure for MLD, and no standard treatment. It is a terminal illness. Children with advanced juvenile or adult onset, and late infantile patients displaying symptoms have treatment limited to pain and symptom management. Presymptomatic late infantile MLD patients, as well as those with juvenile or adult MLD that are either presymptomatic or displaying mild to moderate symptoms, have the option of bone marrow transplantation (including stem cell transplantation), which is under investigation to see if it may slow down progression of disease, or stop its progression in the central nervous system. However, results in the peripheral nervous system have been less dramatic, and the long-term results of these therapies have been mixed.
Several treatment options for the future are currently being investigated.[5] These include gene therapy and enzyme replacement therapy (ERT), substrate reduction therapy (SRT), and potentially enzyme enhancement therapy (EET).
A team of international researchers and foundations organized in 2008 to form an International MLD Registry to create and manage a shared repository of knowledge, including the natural history of MLD. This consortium consists of scientific, academic and industry resources. The registry is not up and operating as of January 2010.
Research Towards a Cure and Clinical Trials
Bone Marrow and Stem Cell Transplant Therapies
- Several trials are underway to continue to improve the effectiveness and reduce the risks of bone marrow and stem cell transplants. Cord blood transplants and reduced preparative routines are being studied.
- Two different approaches to Gene Therapy are being researched for MLD.
- The French group is exploring a direct injection into the brain. They anticipate starting clinical trials in 2012 (current - June 2011)
- Recruiting for the Italian Phase I/II Clinical Trial formally started on March 24, 2010 after approval from the Italian Authorities.The researchers at the San Raffaele Telethon Institute for Gene Therapy in Milan, Italy are testing the efficacy and safety of using an autologous (using the patient's own cells) hematopoietic stem cell transplant (HSCT) to deliver a super-therapeutic ARSA enzyme to the nervous system by the route of the blood cells. Using the patient's own stem cells with genetic correction should reduce or eliminate the complications of graft vs. host disease and provide a long term solution to proper ARSA expression in MLD patients. Bench and animal tests showed positive results.
- Inclusion criteria are pre-symptomatic late infantiles and both pre- and early-symptomatic juveniles. See details on inclusion criteria and the trial protocol here.[6]
- The trial is single center in Milano, Italy. All costs to be paid by the researchers. This is a 3 year study. Four patients have been enrolled and recruiting is still underway.(Current June 2011)
Enzyme replacement therapy (ERT)
- Shire has suspended development of their HGT-1111 intravenous ERT therapy after it was shown to not have sufficient effect. They are resuming work on their internally developed HGT-1110 intrathecal ERT product at the preclinical animal testing stage.
- Work on HGT-1110 was originally stopped when Shire acquired the HGT-1111 product in April 2008. (updated February 2010)[7]
- Shire has orphan status on its HGT-1110 product in both the US and in Europe (9/2010).
- The now canceled HGT-1111 had completed Phase I/II trials in Europe as of September 2008. Initial data looked promising, but data gathered during the post-trial compassionate use/named access period where all patients were put on a uniform higher dose showed no practical benefit.
- Results[8] of the trial were presented at the March 2009 meeting of the ACMG. A video presentation of phase I/II trial summary and a discussion about the phase II/III international clinical trial were presented at the March 2009 Munich and updated at the June 2009 Valley Forge (Philadelphia, PA) MLD Family Conference meetings. Conference videos can be see here.(updated July 2009)
- No published data is available yet on the post-trial study.
- HGT-1111 (formerly called Metazyme) was developed by a Danish company, Zymenex, and was acquired by Shire HGT on April 24, 2008. The product has been granted orphan drug status in the EU and US.
- With the acquisition of HGT-1111 in April 2008, Shire Human Genetic Therapies is expediting this new intravenous ERT therapy in front of its internally developed and now shelved HGT-1110 intrathecal ERT. (updated April 2008)
Substrate reduction therapy
- Zacharon Pharmaceuticals from San Diego is initiating a drug discovery program for MLD. This program is based on using assays which measure sulfatide accumulation in cultured fibroblasts as a means to discover and develop small molecule drugs for MLD. (This approach differs from other approaches which have measured enzyme activity to discover effective drugs.) As of July 2011, Zacharon has begun adapting the assays it developed for other lysosomal storage diseases so that they can be employed to discover and develop drugs for MLD.
- The Cooper Health System (New Jersey) has recently closed enrollment in a clinical trial underway to determine the safety and efficacy of a Vitamin K antagonist (Warfarin) in treating Metachromatic Leukodystrophy (MLD).(current September 2009)
Research & Clinical trial updates provided by the MLD Foundation
See also
MLD Specific Organizations:
Leukodystrophy & Lysosomal Disease Organizations:
- Bethany's Hope (Canada)
- The Stennis Foundation
- ELA, The European Leukodystrophy Association
- The Evanosky Foundation
- Hide & Seek Foundation for Lysosomal Disease Research
- The Myelin Project
External links
- Some portions of this article are courtesy of the public domain text available at the National Institute of Neurological Disorders and Stroke[9]
- Further information regarding MLD, treatments, genetics, and current research projects, can be found at:
- Zacharon Pharmaceuticals
- Shire's drug development pipeline
- 2008 eMedicine article about MLD by Ikeda & Moore of UCLA and Steiner of OHSU
- NIH/GeneReviews at NIH/UW GeneTests overview of MLD written by Arvan Fluharty of UCLA (updated August 2011)
- OMIM entries on ARSA Deficiency
References
- ^ "metachromatic leukodystrophy" at Dorland's Medical Dictionary
- ^ Poeppel P, Habetha M, Marcão A, Büssow H, Berna L, Gieselmann V (March 2005). "Missense mutations as a cause of metachromatic leukodystrophy, Degradation of arylsulfatase A in the endoplasmic reticulum". FEBS J. 272 (5): 1179–88. doi:10.1111/j.1742-4658.2005.04553.x. PMID 15720392.
- ^ Fluharty, Arvan. "Arylsulfatase A Deficiency: Metachromatic Leukodystrophy, ARSA Deficiency". GeneReviews, 2006
- ^ Blomqvist, Maria. Gieselmann,Volkmar. Månsson,Jan-Eric. "Accumulation of lysosulfatide in the brain of arylsulfatase A-deficient mice". Lipids in Health and Disease, 2011
- ^ Biffi A, Lucchini G, Rovelli A, Sessa M (October 2008). "Metachromatic leukodystrophy: an overview of current and prospective treatments". Bone Marrow Transplant. 42 Suppl 2: S2–6. doi:10.1038/bmt.2008.275. PMID 18978739.
- ^ Inclusion criteria, Therapy Description & Contact information
- ^ Shire HGT _statement_2010-02-19.pdf release
- ^ Shire HGT statement
- ^ "NINDS Metachromatic Leukodystrophy Information Page". http://www.ninds.nih.gov/disorders/metachromatic_leukodystrophy/metachromatic_leukodystrophy.htm. Retrieved 2009-06-07.
(LSD) Inborn error of lipid metabolism: lipid storage disorders (E75, 272.7–272.8) Sphingolipidoses
(to ceramide)From globosideGlobotriaosylceramide: Fabry's diseaseFrom sphingomyelinTo sphingosineNCL Other Cerebrotendineous xanthomatosis · Cholesteryl ester storage disease (Lysosomal acid lipase deficiency/Wolman disease) · Sea-blue histiocyte syndromeCategories:- Leukodystrophies
- Lipid storage disorders
- Autosomal recessive disorders
- Rare diseases
- Neurological disorders in children
- Demyelinating diseases of CNS
- If both parents are carriers:

Wikimedia Foundation. 2010.