- Niemann–Pick disease
-
Niemann-Pick disease Classification and external resources ICD-10 E75.2 (ILDS E75.230) ICD-9 272.7 OMIM 257200 607616 257220 607625 DiseasesDB 9016 34341 33390 eMedicine derm/699 MeSH D009542 Niemann–Pick disease (
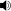 /niːmənˈpɪk/ nee-mən-pik)[1] refers to a group of fatal inherited metabolic disorders that are included in the larger family of lysosomal storage diseases (LSDs).[2]:536
/niːmənˈpɪk/ nee-mən-pik)[1] refers to a group of fatal inherited metabolic disorders that are included in the larger family of lysosomal storage diseases (LSDs).[2]:536Contents
Signs and symptoms
Symptoms are related to the organs in which they accumulate. Enlargement of the liver and spleen (hepatosplenomegaly) may cause reduced appetite, abdominal distension and pain as well as thrombocytopenia secondary to splenomegaly.
Sphingomyelin accumulation in the central nervous system (including the cerebellum) results in unsteady gait (ataxia), slurring of speech (dysarthria) and discoordinated swallowing (dysphagia). Basal ganglia dysfunction causes abnormal posturing of the limbs, trunk and face (dystonia) and upper brainstem disease results in impaired voluntary rapid eye movements (supranuclear gaze palsy). More widespread disease involving the cerebral cortex and subcortical structures is responsible for gradual loss of intellectual abilities causing dementia and seizures.[3]
Bone marrow cavities may be enlarged and the cortical bone may be thinned. Coxa vara may be found.
Sleep related disorders are also seen, including gelastic cataplexy (sudden loss of muscle tone associated with laughter), and sleep inversion (sleepiness during the day and wakefulness at night).
Cause and genetics
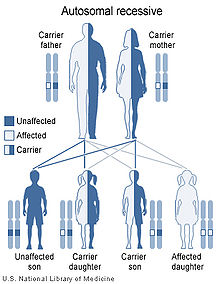 Niemann-Pick disease has an autosomal recessive pattern of inheritance.
Niemann-Pick disease has an autosomal recessive pattern of inheritance.
Mutations in the SMPD1 gene cause Niemann–Pick disease types A and B, and mutations in NPC1 and NPC2 cause Niemann-Pick disease, type C (NPC). Type D was originally separated from Type C to delineate a group of patients with otherwise identical disorders who shared a common Nova Scotian ancestry. Patients in this group are now known to share a specific mutation in the NPC 1 gene, and NPC is now used to embrace both groups. The terms "Niemann-Pick type I" and "Niemann-Pick type II" were proposed to separate the high and low sphingomyelin forms of the disease in the early 1980s, before the molecular defects were described.
Niemann–Pick disease is inherited in an autosomal recessive pattern, which means both copies, or alleles, of the gene must be mutated (altered in such a way that function is impaired, in contrast to a polymorphism, in which the nucleotide sequence is altered but causes no functional disruption) for a person to be affected by the disorder. Most often, the parents of a child with an autosomal recessive disorder are not affected but are carriers of one copy of the altered gene. If both parents are carriers, there is a 25% chance with each pregnancy for an affected child. Genetic counseling and genetic testing is recommended for families who may be carriers of Niemann-Pick.
Classification
In 1961, the following classification was introduced:[4][5]
- Niemann–Pick disease type A: classic infantile
- Niemann–Pick disease type B: visceral
- Niemann-Pick disease, type C: subacute/juvenile
- Niemann–Pick disease type D: Nova Scotian
Now that the genetics are better understood, the condition can be classified as follows:
- Niemann-Pick disease, SMPD1-associated, which includes types A and B
- Niemann-Pick disease, type C, which includes types C1 and C2. (Type D is caused by the same gene as type C1.)
Pathophysiology
Niemann–Pick diseases are genetic diseases which are classified in a subgroup of LSDs called sphingolipidoses or lipid storage disorders in which harmful quantities of fatty substances, or lipids, accumulate in the spleen, liver, lungs, bone marrow, and brain. In the classic infantile type A variant, a missense mutation causes complete deficiency of sphingomyelinase. Sphingomyelin is a component of cell membrane including the organellar membrane and so the enzyme deficiency blocks degradation of lipid, resulting in the accumulation of sphingomyelin within lysosomes in the macrophage-monocyte phagocyte lineage. Affected cells become enlarged, sometimes up to 90 micrometres in diameter, secondary to the distention of lysosomes with sphingomyelin and cholesterol. Histology demonstrates lipid laden macrophages in the marrow, as well as "sea-blue histiocytes" on pathology. Numerous small vacuoles of relatively uniform size are created, imparting a foamy appearance to the cytoplasm.
Treatment
Treatments for Niemann–Pick disease are limited with care being mostly supportive. Anecdotally, organ transplant has been attempted with limited success. Future prospects include enzyme replacement and gene therapy. Bone marrow transplant has been attempted for Type B. Supportive care through nutrition, medication, physical therapy and being followed by specialists can help with quality of life.[3]
In January 2009, Actelion announced that the drug Zavesca (Miglustat) had been approved in the European Union for the treatment of progressive neurological manifestations in adult patients and pediatric patients with Niemann-Pick type C disease (NPC). The drug is available to patients in the United States on an experimental basis. In March 2010, the FDA requested additional preclinical and clinical information regarding Zavesca from Actelion before making a final decision on approving the drug in the United States for NPC disease.
Researchers at the University of Texas Southwestern Medical Center found that when Niemann Pick type C mice were injected with CYCLO (2-hydroxypropyl-β-cyclodextrin or HPBCD), when they were 7 days old, marked improvement in liver function tests, much less neurodegeneration, and, ultimately, significant prolongation of life occurred. These results suggest that 2-hydroxypropyl-β-cyclodextrin acutely reverses the storage defect seen in Niemann-Pick type C disease.[6]
In April 2009, a promising development in treatment of Niemann Pick type C was announced. The U.S. Food and Drug Administration approved Investigational New Drug (INDs) applications for six year old identical twins, Addison and Cassidy Hempel of Reno, Nevada, to receive intravenous infusions of hydroxy-propyl-β-cyclodextrin or HPBCD. The twins are the first in the United States to receive experimental treatment with HPBCD and are currently undergoing treatment at Renown Regional Medical Center in Nevada.
In February 2010, the parents of the twins with the support of Children's Hospital Research Center Oakland, filed an orphan drug application with the Office of Orphan Product Development at the FDA for Trappsol brand cyclodextrin for the treatment of NPC disease. On May 17, 2010, the FDA granted hydroxy-propyl-beta-cyclodextrin orphan drug status and designated hydroxy-propyl-beta-cyclodextrin as a treatment for Niemann Pick Type C disease.
On July 14, 2010, Children's Hospital Research Center Oakland filed new Investigational New Drug (INDs) applications with the FDA on behalf of the Hempel twins based on promising new animal data. Doctors have requested to deliver hydroxy-propyl-beta-cyclodextrin intrathecally and directly into the central nervous system of the twins in an attempt to arrest neurodegeneration. The INDs were approved on September 23, 2010, and treatments are expected to start on October 15, 2010.[citation needed]
Prognosis
Type A Niemann Pick disease has an extremely poor prognosis with most cases being fatal by the age of 18 months.[7] Type B and C Niemann Pick disease have a better prognosis, with many patients with these disorders living into their teens or adulthood.[8]
History
Albert Niemann published the first description of what is now known as Niemann–Pick disease, type A, in 1914. Ludwig Pick described the pathology of the disease in a series of papers in the 1930s.[9][10][11]
See also
- Gaucher's disease
- Medical genetics of Ashkenazi Jews
References
- ^ "Niemann–Pick". Oxford English Dictionary. Oxford University Press. 2nd ed. 1989.
- ^ James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. ISBN 0-7216-2921-0.
- ^ a b Emedicine - Niemann-Pick disease Treatment & Medication, http://emedicine.medscape.com/article/951564-treatment
- ^ Crocker AC (April 1961). "The cerebral defect in Tay-Sachs disease and Niemann-Pick disease". Journal of neurochemistry 7: 69–80. doi:10.1111/j.1471-4159.1961.tb13499.x. PMID 13696518.
- ^ "OMIM - NIEMANN-PICK DISEASE, TYPE C1; NPC1". http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=257220. Retrieved 2008-10-27.
- ^ Liu, Benny; et al. (2009). "Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1−/− mouse". PNAS 106 (7): 2377–2382. doi:10.1073/pnas.0810895106. Cited in Chemical & Engineering News, 9 February 2009 p. 9
- ^ http://www.ninds.nih.gov/disorders/niemann/niemann.htm
- ^ http://ghr.nlm.nih.gov/condition=niemannpickdisease
- ^ synd/1029 at Who Named It?
- ^ Niemann, A. (1914). "Ein unbekanntes Krankheitsbild". Jahrbuch für Kinderheilkunde. Neue Folge (Berlin) 79: 1–10.
- ^ Pick, L. (1926). "Der Morbus Gaucher und die ihm ähnlichen Krankheiten (die lipoidzellige Splenohepatomegalie Typus Niemann und die diabetische Lipoidzellenhypoplasie der Milz)". Ergebnisse der Inneren Medizin und Kinderheilkunde (Berlin) 29: 519–627.
External links
- National Niemann-Pick Disease Foundation (U.S.)
- Ara Parseghian Medical Research Foundation
- Niemann-Pick Disease Group (UK)
- GeneReviews/NCBI/NIH/UW entry on Acid Sphingomyelinase Deficiency Includes: Niemann-Pick Disease Type A, Niemann-Pick Disease Type B
- OMIM entries on Acid Sphingomyelinase Deficiency
- GeneReviews/NCBI/NIH/UW entry on Niemann-Pick Disease Type C
- OMIM entries on Niemann-Pick Type C
- Niemann-Pick Disease Group Canada
- National Institutes of Health Clinical Center Study On Niemann Pick Type C
- Marc C. Patterson, MD, child neurologist, Mayo Clinic
- National Institute of Neurological Diseaases and Strokes
- Addi and Cassi Hempel: Identical twins with Niemann Pick type C disease being treated with cyclodextrin
- Genetics Home Reference on Niemann Pick Disease
- Hide & Seek Foundation for Lysosomal Disease Research
- Detailed information about Niemann Pick Type C for patients and Healthcare Professionals
- Pathology images and digital slides (HP:7983)
- This article incorporates public domain text from The U.S. National Library of Medicine
(LSD) Inborn error of lipid metabolism: lipid storage disorders (E75, 272.7–272.8) Sphingolipidoses
(to ceramide)From globosideGlobotriaosylceramide: Fabry's diseaseFrom sphingomyelinTo sphingosineNCL Other Cerebrotendineous xanthomatosis · Cholesteryl ester storage disease (Lysosomal acid lipase deficiency/Wolman disease) · Sea-blue histiocyte syndromeHistiocytosis (D76.0, 277.89) WHO-I/Langerhans cell histiocytosis/
X-type histiocytosisLetterer–Siwe disease · Hand–Schüller–Christian disease · Eosinophilic granuloma · Congenital self-healing reticulohistiocytosisWHO-II/non-Langerhans cell histiocytosis/
Non-X histiocytosisJuvenile xanthogranuloma · Hemophagocytic lymphohistiocytosis · Erdheim-Chester disease · Niemann-Pick disease · Sea-blue histiocyte syndrome · Benign cephalic histiocytosis · Generalized eruptive histiocytoma · Xanthoma disseminatum · Progressive nodular histiocytosis · Papular xanthoma · Hereditary progressive mucinous histiocytosis · Reticulohistiocytosis (Multicentric reticulohistiocytosis, Reticulohistiocytoma) · Indeterminate cell histiocytosisWHO-III/malignant histiocytosis Histiocytic sarcoma · Langerhans cell sarcoma · Interdigitating dendritic cell sarcoma · Follicular dendritic cell sarcomaUngrouped Categories:- Autosomal recessive disorders
- Rare diseases
- Lipid storage disorders
- Ashkenazi Jews topics
- Skin conditions resulting from errors in metabolism
- Neurodegenerative disorders
Wikimedia Foundation. 2010.