- Langerhans cell histiocytosis
-
Langerhans cell histiocytosis Classification and external resources 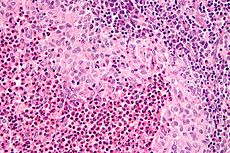
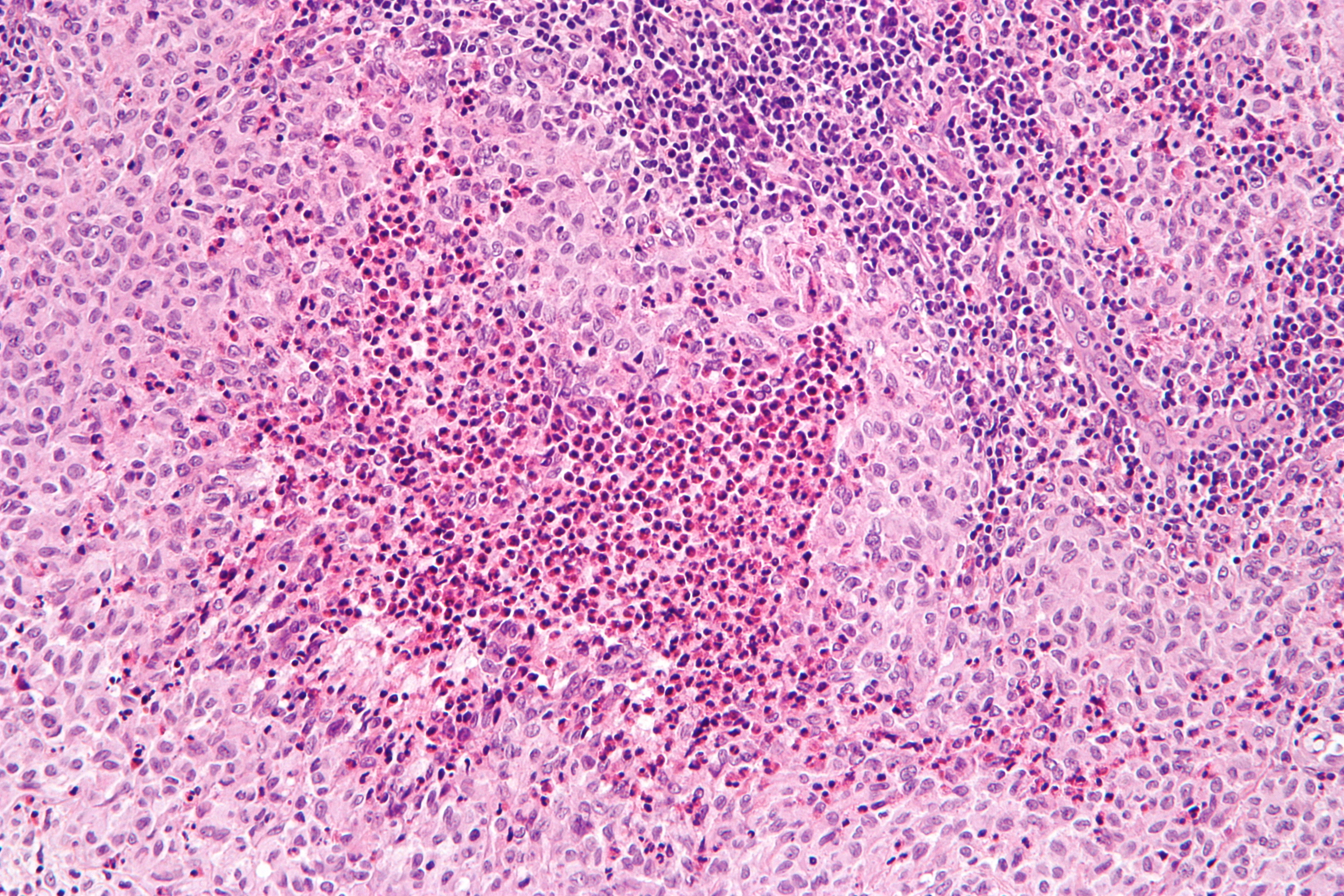
Micrograph showing a Langerhans cell histiocytosis with the characteristic reniform Langerhans cells accompanied by abundant eosinophils. H&E stain.ICD-10 D76.0 ICD-9 202.5, 277.89, 516.5 ICD-O: M9752/3,M9751/1 OMIM 604856 DiseasesDB 5906 eMedicine derm/216 MeSH D006646 Langerhans cell Histiocytosis (LCH) is a rare disease involving clonal proliferation of Langerhans cells, abnormal cells deriving from bone marrow and capable of migrating from skin to lymph nodes. Clinically, its manifestations range from isolated bone lesions to multisystem disease.
LCH is part of a group of clinical syndromes called histiocytoses, which are characterized by an abnormal proliferation of histiocytes (an archaic term for activated dendritic cells and macrophages). These diseases are related to other forms of abnormal proliferation of white blood cells, such as leukemias and lymphomas.
The disease has gone by several names, including Hand-Schüller-Christian disease, Abt-Letterer-Siwe disease, and histiocytosis X, until it was renamed in 1985 by the Histiocyte Society.[1]
Contents
Classification
Alternative names Histiocytosis X Histiocytosis X syndrome
Subordinate terms Hand-Schüller-Christian disease Letterer-Siwe disease
Histiocytosis X, unspecified
Eosinophilic granulomatosis
Langerhans cell granulomatosis
Langerhans cell histiocytosis, Hashimoto-Pritzker type
Langerhans cell histiocytosis of lung
Langerhans cell histiocytosis, disseminated (clinical)
Langerhans cell histiocytosis, unifocal (clinical)The disease spectrum results from clonal accumulation and proliferation of cells resembling the epidermal dendritic cells called Langerhans cells, hence sometimes called dendritic cell histiocytosis. These cells in combination with lymphocytes, eosinophils, and normal histiocytes form typical LCH lesions that can be found in almost any organ.[2]
There are three types of histiocytoses: malignant (true histiocytic lymphomas), "reactive" (benign histiocytoses), Langerhans cell histiocytosis.[3]
"Reactive" in this context indicates that the abnormality may be due to a physiological reaction to infection. For example leukocytosis (proliferation of white blood cells) is a normal reaction to infection, and "histiocytes" are developmentally related to white blood cells.
LCH is traditionally divided into three groups: unifocal, multifocal unisystem, and multifocal multisystem.[3]
- Unifocal
- Unifocal LCH, also called eosinophilic granuloma (an older term which is now known to be a misnomer), is a slowly-progressing disease characterized by an expanding proliferation of Langerhans cells in various bones. It is a monostotic or polystotic disease with no extraskeletal involvement. This differentiates eosinofilic granuloma from other forms of langerhans cell histiocytosis (Letterer-Siwe or Hand-Schüller-Christian variant.[4]
- Multifocal unisystem
- Seen mostly in children, multifocal unisystem LCH is characterized by fever, bone lesions and diffuse eruptions, usually on the scalp and in the ear canals. 50% of cases involve the pituitary stalk, leading to diabetes insipidus. The triad of diabetes insipidus, exopthalmos, and lytic bone lesions is known as the Hand-Schüller-Christian triad.
- Multifocal multisystem
- Multifocal multisystem LCH, also called Letterer-Siwe disease, is a rapidly-progressing disease in which Langerhans cells proliferate in many tissues. It is mostly seen in children under age 2, and the prognosis is poor: even with aggressive chemotherapy, the 5-year survival is only 50%.[5]
Pulmonary Langerhans cell histiocytosis (PLCH) is a unique form of LCH in that it occurs almost exclusively in cigarette smokers. It is now considered a form of smoking-related interstitial lung disease. Some patients recover completely after they stop smoking, but others develop long-term complications such as pulmonary fibrosis and pulmonary hypertension.
Prevalence
LCH usually affects children between 1 and 15 years old, with a peak incidence between 5 and 10 years of age. Among children under the age of 10, yearly incidence is thought to be 1 in 200,000;[6] and in adults even rarer, in about 1 in 560,000.[7] It has been reported in elderly but is vanishingly rare.[8] It is most prevalent in Caucasians, and affects males twice as often as females.[citation needed]
LCH is usually a sporadic and non-hereditary condition but familial clustering has been noted in limited number of cases. Hashimoto-Pritzker disease is a congenital self-healing variant of Hand-Schüller-Christian disease.[9]
Signs and symptoms
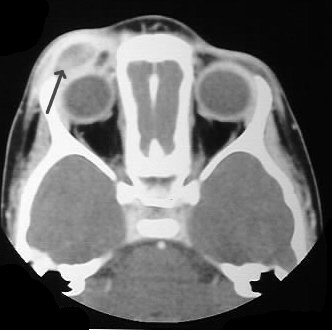
 CT scan showing LCH infiltrating peri-orbital tissue (arrowed).
CT scan showing LCH infiltrating peri-orbital tissue (arrowed).
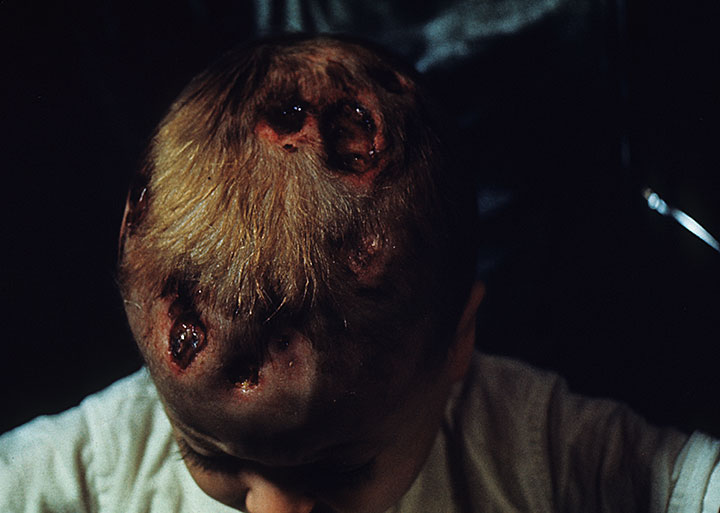
 A patient with Hand–Schüller–Christian disease which is a subtype of Langerhans cell histiocytosis.
A patient with Hand–Schüller–Christian disease which is a subtype of Langerhans cell histiocytosis.LCH provokes a non-specific inflammatory response, which includes fever, lethargy, and weight loss. Organ involvement can also cause more specific symptoms.
- Bone: The most-frequently seen symptom in both unifocal and multifocal disease is painful bone swelling. The skull is most frequently affected, followed by the long bones of the upper extremities and flat bones. Infiltration in hands and feet is unusual. Osteolytic lesions can lead to pathological fractures.
- Skin: Commonly seen are a rash which varies from scaly erythematous lesions to red papules pronounced in intertriginous areas. Up to 80% of LCH patients have extensive eruptions on the scalp.
- Bone marrow: Pancytopenia with superadded infection usually implies a poor prognosis. Anemia can be due to a number of factors and does not necessarily imply bone marrow infiltration.
- Lymph node: Enlargement of the liver in 20%, spleen in 30% and lymph nodes in 50% of histiocytosis cases.[10]
- Endocrine glands: Hypothalamic pituitary axis commonly involved. Diabetes insipidus is most common. Anterior pituitary hormone deficiency is usually permanent.
- Lungs: some patients are asymptomatic, diagnosed incidentally because of lung nodules on radiographs; others suffer from chronic cough and shortness of breath.
- Less frequently gastrointestinal tract and central nervous system.
Diagnosis
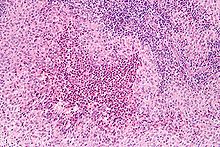
Diagnosis is confirmed histologically by tissue biopsy. Hematoxylin-eosin stain of biopsy slide will show features of Langerhans cell e.g. distinct cell margin, pink granular cytoplasm. Presence of Birbeck granules on electron microscopy and immuno-cytochemical features e. g. CD1 positivity are more specific. Initially routine blood tests e.g. full blood count, liver function test, U&Es, bone profile are done to determine disease extent and rule out other causes. Radiology will show osteolytic bone lesions and damage to the lung. The latter may be evident in chest X-rays with micronodular and interstitial[disambiguation needed
 ] infiltrate in the mid and lower zone of lung, with sparing of the Costophrenic angle or honeycomb appearance in older lesions. MRI and CT may show infiltration in sella turcica. Assessment of endocrine function and bonemarrow biopsy are also performed when indicated.
] infiltrate in the mid and lower zone of lung, with sparing of the Costophrenic angle or honeycomb appearance in older lesions. MRI and CT may show infiltration in sella turcica. Assessment of endocrine function and bonemarrow biopsy are also performed when indicated.Treatment
Treatment is guided by extent of disease. Solitary bone lesion may be amenable through excision or limited radiation. However systemic diseases often require chemotherapy. Use of systemic steroid is common, singly or adjunct to chemotherapy. Local steroid cream is applied to skin lesions. Endocrine deficiency often require lifelong supplement e.g. desmopressin for diabetes insipidus which can be applied as nasal drop. Chemotherapeutic agents such as alkylating agents, antimetabolites, vinca alkaloids either singly or in combination can lead to complete remission in diffuse disease.
Prognosis
Excellent for single foci disease. With multi-focal disease 60% have a chronic course, 30% achieve remission and mortality is up to 10%.[11]
See also
- X-type histiocytosis
References
- ^ "Histiocytosis syndromes in children. Writing Group of the Histiocyte Society". Lancet 1 (8526): 208–9. 1987. doi:10.1016/S0140-6736(02)95528-5. PMID 2880029.
- ^ Makras P, Papadogias D, Kontogeorgos G, Piaditis G, Kaltsas G (2005). "Spontaneous gonadotrophin deficiency recovery in an adult patient with Langerhans cell histiocytosis (LCH)". Pituitary 8 (2): 169–74. doi:10.1007/s11102-005-4537-z. PMID 16379033.
- ^ a b Cotran, Ramzi S.; Kumar, Vinay; Abbas, Abul K.; Fausto, Nelson; Robbins, Stanley L. (2005). Robbins and Cotran pathologic basis of disease. St. Louis, Mo: Elsevier Saunders. pp. 701-. ISBN 0-8089-2302-1.
- ^ Template:See chapter 501 in Nelson Textbook of Pediatrics 18th ed. Question 45 same chapter
- ^ http://emedicine.medscape.com/article/1100579-overview
- ^ "MedlinePlus Medical Encyclopedia: Histiocytosis". http://www.nlm.nih.gov/medlineplus/ency/article/000068.htm. Retrieved 2007-05-10.
- ^ "Histiocytosis Association of Canada". http://www.histiocytosis.ca/facts.html. Retrieved 2007-05-16.
- ^ Gerlach B, Stein A, Fischer R, Wozel G, Dittert D, Richter G (1998). "[Langerhans cell histiocytosis in the elderly]" (in German). Der Hautarzt; Zeitschrift für Dermatologie, Venerologie, und verwandte Gebiete 49 (1): 23–30. PMID 9522189.
- ^ Kapur P, Erickson C, Rakheja D, Carder K, Hoang M (2007). "Congenital self-healing reticulohistiocytosis (Hashimoto-Pritzker disease): ten-year experience at Dallas Children's Medical Center". J. Am. Acad. Dermatol. 56 (2): 290–4. doi:10.1016/j.jaad.2006.09.001. PMID 17224372.
- ^ "Langerhans cell histiocytosis - Patient UK". http://www.patient.co.uk/showdoc/40002781/#notes. Retrieved 2007-05-10.
- ^ Komp D, El Mahdi A, Starling K, Easley J, Vietti T, Berry D, George S (1980). "Quality of survival in histiocytosis X: a Southwest Oncology Group study". Med. Pediatr. Oncol. 8 (1): 35–40. doi:10.1002/mpo.2950080106. PMID 6969347.
External links
- [1] a EU network granted by the EU commission with a multilanguage web site
- Images of LCH MedPix(r)Database
- Histiocyte Society An International Community Dedicated to Research and Treatment
- Seven Part Video Series
- Langerhans cell histiocytosis, article by the Sydney Children's Hospital
- 5-Minute Clinical Consult
- synd/2224 at Who Named It?
Histiocytosis (D76.0, 277.89) WHO-I/Langerhans cell histiocytosis/
X-type histiocytosisLetterer–Siwe disease · Hand–Schüller–Christian disease · Eosinophilic granuloma · Congenital self-healing reticulohistiocytosisWHO-II/non-Langerhans cell histiocytosis/
Non-X histiocytosisJuvenile xanthogranuloma · Hemophagocytic lymphohistiocytosis · Erdheim-Chester disease · Niemann-Pick disease · Sea-blue histiocyte syndrome · Benign cephalic histiocytosis · Generalized eruptive histiocytoma · Xanthoma disseminatum · Progressive nodular histiocytosis · Papular xanthoma · Hereditary progressive mucinous histiocytosis · Reticulohistiocytosis (Multicentric reticulohistiocytosis, Reticulohistiocytoma) · Indeterminate cell histiocytosisWHO-III/malignant histiocytosis Histiocytic sarcoma · Langerhans cell sarcoma · Interdigitating dendritic cell sarcoma · Follicular dendritic cell sarcomaUngrouped Categories:- Histiocytosis
- Monocyte- and macrophage-related cutaneous conditions
- Rare diseases

Wikimedia Foundation. 2010.