- Porphyria cutanea tarda
DiseaseDisorder infobox
Name = Porphyria cutanea tarda
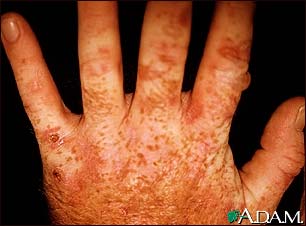
Caption = Porphyria cutanea tarda on the hands
ICD10 = ICD10|E|80|1|e|70
ICD9 = ICD9|277.1Porphyria cutanea tarda (PCT) is the most common subtype of
porphyria . The disorder results from low levels of theenzyme responsible for the fifth step inheme production. Heme is a vital molecule for all of the body's organs. It is a component ofhemoglobin , the molecule that carriesoxygen in the blood.Presentation
Porphyria cutanea tarda (commonly referred to as PCT) is recognized as the most prevalent subtype of porphyritic diseases. The disease is characterized by blistering of the skin in areas that receive higher levels of exposure to sunlight. The primary cause of this disorder is a deficiency of uroporphyrinogen decarboxylase (UROD), a cytosolic enzyme that is a step in the enzymatic pathway that leads to the synthesis of heme.1 While a deficiency in this enzyme is the direct cause leading to this disorder, there are a number of both genetic and environmental risk factors that are associated with PCT. [cite journal | author=Kushner, J; et. al | title=An inherited enzymatic defect in porphyria cutanea tarda: decreased uroporphyrinogen decarboxylase activity | journal=J Clin Invest | year=1976 | pages=1089–97 | volume=58 | issue=5 | pmid=11682034 | doi=10.1172/JCI108560]
igns and Symptoms
Typically, patients who are ultimately diagnosed with PCT first seek treatment following the development photosensitivities in the form of
blisters and erosions on commonly exposed areas of theskin . This is usually observed in the face, hands, forearms, and lower legs. It heals slowly and withscarring . Though blisters are the most common skin manifestations of PCT, other skin manifestations likehyperpigmentation (as if they are getting a tan) andhypertrichosis (mainly on top of the cheeks) also occur. PCT is a chronic condition, with external symptoms often subsiding and recurring as a result of a number of factors. In addition to the symptomatic manifestation of the disease in the skin, chronicliver problems are extremely common in patients with thesporadic form of PCT. These include hepatic fibrosis (scarring of the liver),cirrhosis , andinflammation . However, liver problems are less common in patients with theinherited form of thedisease . [cite journal | author=DiPadova, C; et. al | title=Effects of phlebotomy on urinary porphyrin pattern and liver histology in patients with porphyria cutanea tarda | journal=Am J Med Sci | year=1983 | pages=2–12 | volume=285 | issue=1 | pmid=11682034 | doi=10.1097/00000441-198301000-00001]Epidemiology
This type of porphyria occurs in an estimated 1 in 25,000 people, including both inherited and sporadic (noninherited) cases. An estimated 80 % of porphyria cutanea tarda cases are sporadic. The exact frequency is not clear because many people with the condition never experience symptoms.
Genetics
Inherited mutations in the "
UROD "gene cause about 20 % of cases (the other 80 % of cases do not havemutation s in UROD, and are classified as sporadic). UROD makes an enzyme calleduroporphyrinogen III decarboxylase , which is critical to the chemical process that leads to heme production. The activity of this enzyme is usually reduced by 50 % in all tissues in people with the inherited form of the condition.Nongenetic factors such as
alcohol abuse , excessiron , and others listed above can increase the demand for heme and the enzymes required to make heme. The combination of this increased demand and reduced activity of uroporphyrinogen decarboxylase disrupts heme production and allows byproducts of the process to accumulate in the body, triggering the signs and symptoms of porphyria cutanea tarda.The "
HFE " gene makes a protein that helps cells regulate the absorption of iron from the digestive tract and into the cells of the body. Certain mutations in the "HFE" gene causehemochromatosis (aniron overload disorder ). People who have these mutations are also at an increased risk of developing porphyria cutanea tarda.In the 20% of cases where porphyria cutanea tarda is inherited, it is inherited in an
autosomal dominant pattern, which means one copy of the altered gene is sufficient to decrease enzyme activity and cause the signs and symptoms of the disorder.Risk Factors
While inherited deficiencies in uroporphyrinogen decarboxylase often lead to the development of PCT, there are a number of risk factors that can both cause and exacerbate the symptoms of this disease. One of the most common risk factors observed is infection with the Hepatitis C virus. One review of a collection of PCT studies noted Hepatitis C infection in 50% of documented cases of PCT. Additional risk factors include alcohol abuse, excess iron, and exposure to chlorinated cyclic hydrocarbons.
Pathogenesis
As stated previously, porphyria cutanea tarda is primarily caused by uroporphyrinogen decarboxylase deficiency (UROD). UROD occurs in nature as a homodimer of two subunits. It is the fifth step in heme synthesis pathway, and is active in the cytosol. This enzymatic conversion results in coproporphyrinogen III as the primary product. This is accomplished by the clockwise removal of the four carboxyl groups present in the cyclic uroporphyrinogen III molecule. Therefore, a deficiency in this enzyme causes the aforementioned buildup of uroporphyrinogen in the urine, which can be helpful in the diagnosis of this disorder.
The dermatological symptoms of PCT that include blistering and lesions on sun-exposed areas of the skin are caused by a buildup of porphyrin compounds (specifically uroporphyrinogen) close to the surface of the skin. Due to the highly conjugated structure of porphyrins involving alternating single and double carbon bonds, these compounds exhibit a deep purple color, resulting in the discoloration observed in the skin. The excess uroporphyrinogen that lead to these lesions is primarily produced in the liver, but exposure to excess sunlight is thought to lead to the production of reactive oxygen species, disrupting the activity of uroporphyrinogen decarboxylase and contributing to the buildup of uroporphyrinogen. This is thought to be the underlying biochemical pathway behind the photosensitivity observed in patients with PCT.The strong association of PCT with Hepatitis C infection is not entirely understood. Studies have suggested that the cytopathic effect of the virus on hepatocytes can lead to the release of free iron. This iron can disrupt the activity of cytochrome p450, releasing activated oxygen species. These can oxidize the UROD substrate uroporphyrinogen, which can result in the inhibition of UROD and lead to deficient activity of this key enzyme.
Excess alcohol abuse is frequently associated with both inducing PCT and aggravating a preexisting diagnosis of the disorder. It is thought to do so by causing oxidative damage to liver cells, resulting in oxidized species of uroporphyrinogen that inhibit the activity of hepatic UROD. It is also felt to increase the uptake of iron in liver cells, leading to further oxidation of uroporphyrinogen by the release of activated oxygen species. Additionally, exposure to chlorinated cyclic hydrocarbons can lead to a deficiency in the activity of uroporphyrinogen decarboxylase, causing the buildup of excess uroporphyrinogen.
Types
Type I porphyria cutanea tarda
Type I porphyria cutanea tarda, the sporadic form, is indicated by UROD deficiency that is observed only in hepatic cells and nowhere else in the body. Genetically, these individuals will not exhibit deficiency in the UROD gene, although other genetic factors such as HFE deficiency (resulting in hemochromatosis and the buildup of iron in the liver) are thought to play a key role. Typically in these individuals, a variety of risk factors such as alcohol abuse and Hepatitis C infection conspire to result in the clinical manifestation of PCT.
Type II porphyria cutanea tarda
Patients exhibiting Type II PCT have a specific deficiency in the UROD gene, passed down in an autosomal dominant pattern. Those possessing this deficiency are heterozygous for the UROD gene. They do not show a complete lack of functional uroporphyrinogen decarboxylase, only a deficient form of the enzyme that is marked by reduced conversion of uroporphyrinogen to coproporphyrinogen. Therefore, the expression of uroporphyrinogen decarboxylase will be reduced throughout the body of these individuals, while it is isolated to the liver in Type I patients. While this genetic deficiency is the main distinction between Type I and Type II PCT, the risk factors mentioned before are often seen in patients presenting with Type II PCT. In fact, many people who possess the deficient UROD gene often go their entire lives without having a clinical manifestation of PCT symptoms.
Type III porphyria cutanea tarda
The least common is Type III, which is no different from Type I insofar as the patients possess normal UROD genes. Despite this, Type III PCT is observed in more than one family member, indicating a genetic component unrelated to the expression of uroporphyrinogen decarboxylase.
Precipitating factors
* Alcohol
* Estrogen
* IronDiagnosis
While the most common symptom of PCT is the appearance of skin lesions and blistering, their appearance does not single-handedly lead to a conclusive diagnosis. Laboratory testing will commonly reveal high levels of uroporphyrinogen in the urine, clinically referred to as uroporphyrinogenuria. Additionally, testing for common risk factors such as Hepatitis C and hemochromatosis is strongly suggested, as their high prevalence in patients with PCT may require additional treatment.
Treatment
Since PCT is a chronic condition, a comprehensive management of the disease is the most effective means of treatment. Primarily, it is key that patients diagnosed with PCT avoid alcohol consumption, iron supplements, excess exposure to sunlight (especially in the summer), as well as estrogen and chlorinated cyclic hydrocarbons, all of which can potentially exacerbate the disorder. Additionally, the management of excess iron (due to the commonality of hemochromatosis in PCT patients) can be achieved through
phlebotomy , whereby blood is systematically drained from the patient. Finally, due to the strong association between PCT and Hepatitis C, the treatment of Hepatitis C (if present) is vital to the effective treatment of PCT.Chloroquine andvenesection can be part of a management strategy. [cite journal | author=Sarkany RP | title=The management of porphyria cutanea tarda | journal=Clin Exp Dermatol | year=2001 | pages=225–32 | volume=26 | issue=3 | pmid=11422163 | doi=10.1046/j.1365-2230.2001.00825.x]Cultural references
Porphyria Cutanea Tarda is also the name of a song by the punk band AFI on their album
Black Sails in the Sunset .References
*
*
*
*
*.
*
*
*
*
*
*
* [http://web.uct.ac.za/depts/porphyria/professional/prof-pct.htm "Porphyria Cutanea Tarda"] Porphyria South Africa, University of Cape Town/Groote Schurr Hospital
*RareDiseases|7433|Porphyria cutanea tarda
Wikimedia Foundation. 2010.