- Disseminated intravascular coagulation
-
Disseminated intravascular coagulation or Disseminated intravascular coagulopathy Classification and external resources 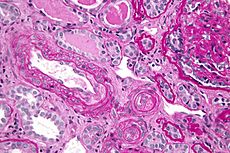
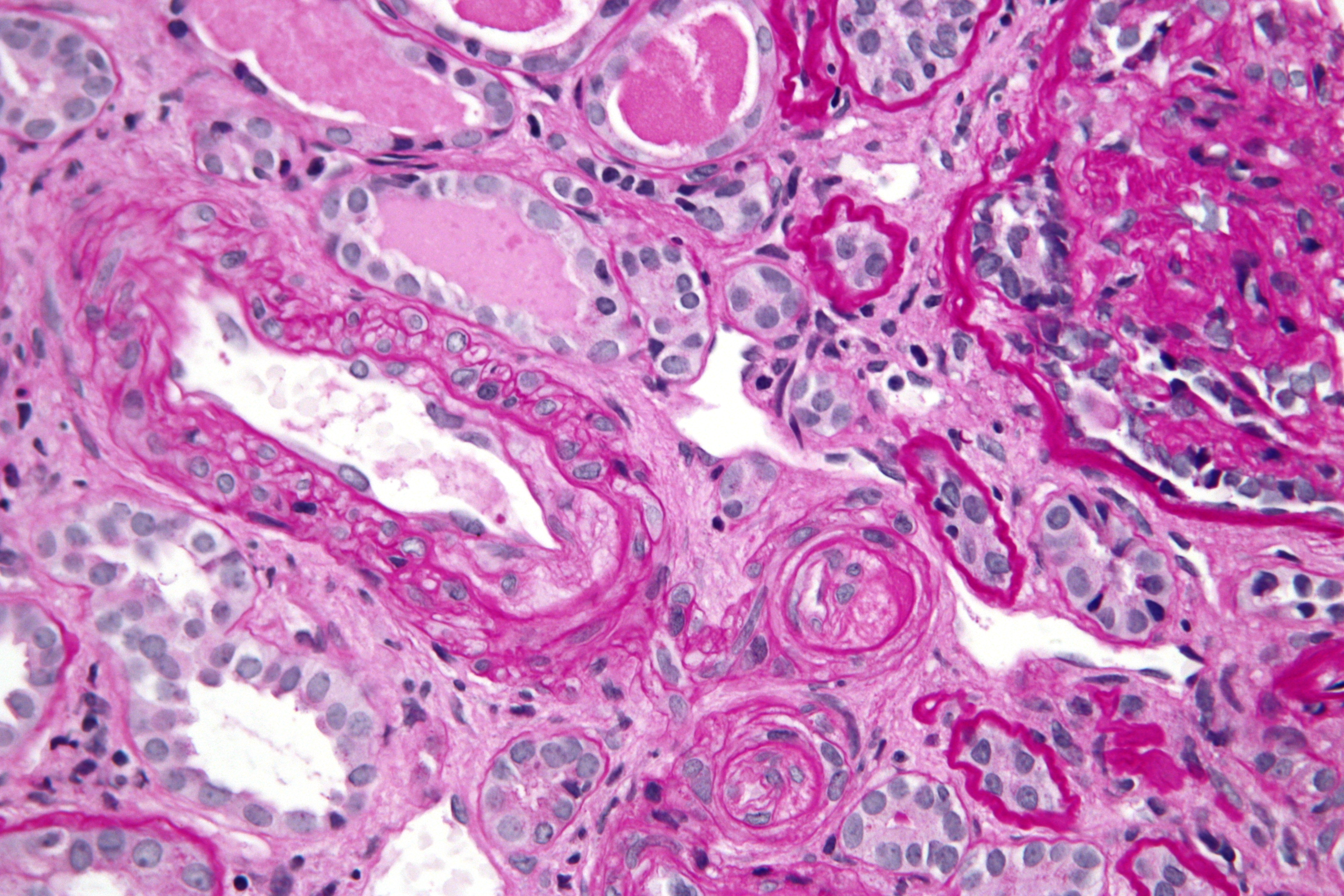
Micrograph showing a thrombotic microangiopathy, as may be seen in DIC. Kidney biopsy. PAS stain.ICD-10 D65 ICD-9 286.6 DiseasesDB 3765 eMedicine med/577 emerg/150 MeSH D004211 Disseminated intravascular coagulation (DIC), also known as disseminated intravascular coagulopathy or consumptive coagulopathy, is a pathological activation of coagulation (blood clotting) mechanisms that happens in response to a variety of diseases. DIC leads to the formation of small blood clots inside the blood vessels throughout the body.[1] As the small clots consume coagulation proteins and platelets, normal coagulation is disrupted and abnormal bleeding occurs from the skin (e.g. from sites where blood samples were taken), the gastrointestinal tract, the respiratory tract and surgical wounds. The small clots also disrupt normal blood flow to organs (such as the kidneys), which may malfunction as a result.[2]
DIC can occur acutely but also on a slower, chronic basis, depending on the underlying problem.[3] It is common in the critically ill, and may participate in the development of multiple organ failure, which may lead to death.[4]
Contents
Pathophysiology
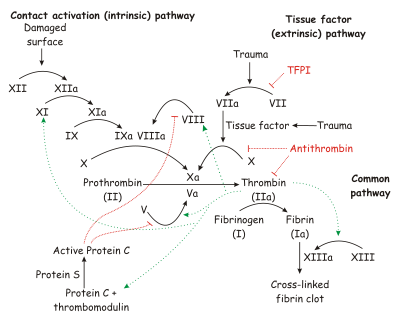 The coagulation cascade of secondary hemostasis.
The coagulation cascade of secondary hemostasis.
Under homeostatic conditions, the body is maintained in a finely tuned balance of coagulation and fibrinolysis. The activation of the coagulation cascade yields thrombin that converts fibrinogen to fibrin; the stable fibrin clot being the final product of hemostasis. The fibrinolytic system then functions to break down fibrinogen and fibrin. Activation of the fibrinolytic system generates plasmin (in the presence of thrombin), which is responsible for the lysis of fibrin clots. The breakdown of fibrinogen and fibrin results in polypeptides called fibrin degradation products (FDPs) or fibrin split products (FSPs). In a state of homeostasis, the presence of plasmin is critical, as it is the central proteolytic enzyme of coagulation and is also necessary for the breakdown of clots, or fibrinolysis.
In DIC, the processes of coagulation and fibrinolysis are dysregulated, and the result is widespread clotting with resultant bleeding. Regardless of the triggering event of DIC, once initiated, the pathophysiology of DIC is similar in all conditions. One critical mediator of DIC is the release of a transmembrane glycoprotein called tissue factor (TF). TF is present on the surface of many cell types (including endothelial cells, macrophages, and monocytes) and is not normally in contact with the general circulation, but is exposed to the circulation after vascular damage. For example, TF is released in response to exposure to cytokines (particularly interleukin 1), tumor necrosis factor, and endotoxin.[5] This plays a major role in the development of DIC in septic conditions. TF is also abundant in tissues of the lungs, brain, and placenta. This helps to explain why DIC readily develops in patients with extensive trauma. Upon activation, TF binds with coagulation factors that then triggers the extrinsic pathway (via Factor VII) which subsequently triggers the intrinsic pathway (XII to XI to IX) of coagulation.
The release of endotoxin is the mechanism by which Gram-negative sepsis provokes DIC. In acute promyelocytic leukemia, treatment causes the destruction of leukemic granulocyte precursors, resulting in the release of large amounts of proteolytic enzymes from their storage granules, causing microvascular damage. Other malignancies may enhance the expression of various oncogenes that result in the release of TF and plasminogen activator inhibitor-1 (PAI-1), which prevents fibrinolysis.[6]
Excess circulating thrombin results from the excess activation of the coagulation cascade. The excess thrombin cleaves fibrinogen, which ultimately leaves behind multiple fibrin clots in the circulation. These excess clots trap platelets to become larger clots, which leads to microvascular and macrovascular thrombosis. This lodging of clots in the microcirculation, in the large vessels, and in the organs is what leads to the ischemia, impaired organ perfusion, and end-organ damage that occurs with DIC.
Coagulation inhibitors are also consumed in this process. Decreased inhibitor levels will permit more clotting so that a feedback system develops in which increased clotting leads to more clotting. At the same time, thrombocytopenia occurs and this has been attributed to the entrapment and consumption of platelets. Clotting factors are consumed in the development of multiple clots, which contributes to the bleeding seen with DIC.
Simultaneously, excess circulating thrombin assists in the conversion of plasminogen to plasmin, resulting in fibrinolysis. The breakdown of clots results in excess amounts of FDPs, which have powerful anticoagulant properties, contributing to hemorrhage. The excess plasmin also activates the complement and kinin systems. Activation of these systems leads to many of the clinical symptoms that patients experiencing DIC exhibit, such as shock, hypotension, and increased vascular permeability. The acute form of DIC is considered an extreme expression of the intravascular coagulation process with a complete breakdown of the normal homeostatic boundaries. DIC is associated with a poor prognosis and a high mortality rate.
There has been a recent challenge however to the basic assumptions and interpretations of the pathophysiology of DIC. A study of sepsis and DIC in animal models has shown that a highly-expressed receptor on the surface of hepatocytes, termed the Ashwell-Morell receptor, is responsible for thrombocytopenia in bacteremia and sepsis due to streptococcal pneumoniae (SPN) and possibly other pathogens. The thrombocytopenia observed in SPN sepsis was not due to increased consumption of coagulation factors such as platelets, but instead was the result of this receptor's activity enabling hepatocytes to ingest and rapidly clear platelets from circulation.[7] By removing pro-thrombotic components before they participate in the coagulopathy of DIC, the Ashwell-Morell receptor lessens the severity of DIC, reducing thrombosis and tissue necrosis, and promoting survival. The hemorrhage observed in DIC and among some tissues lacking this receptor may thereby be secondary to increased thrombosis with loss of the mechanical vascular barrier. This discovery has possible significant clinical implications in devising new approaches to reducing the pathophysiology of DIC.
Causes
DIC can occur in the following conditions:[3][4][8]
- Cancers of lung, pancreas, prostate and stomach, as well as acute myeloid leukemia (particularly APML)
- Obstetric: abruptio placentae, pre-eclampsia, amniotic fluid embolism
- Massive tissue injury: Trauma, burns, extensive surgery
- Infections: Gram-negative sepsis, Neisseria meningitidis, Streptococcus pneumoniae, malaria, histoplasmosis, aspergillosis, Rocky mountain spotted fever
- Miscellaneous: Liver disease, snake bite, giant hemangioma, shock, heat stroke, vasculitis, aortic aneurysm, Serotonin syndrome[9]
- Viral: Arenaviruses causing Argentine hemorrhagic fever or Bolivian Hemorrhagic Fever
Signs and symptoms
The affected person is often acutely ill and shocked with widespread haemorrhage (common bleeding sites are mouth, nose and venepuncture sites), extensive bruising, renal failure and gangrene.[8][10] The onset of DIC can be fulminant, as in endotoxic shock or amnioitic fluid embolism, or it may be insidious and chronic, as in cases of carcinomatosis.[3]
Diagnosis
Diagnosis is usually suggested by following conditions:[8]
- Severe cases with hemorrhage: The PT and APTT are usually very prolonged and the fibrinogen level markedly reduced. High levels of fibrin degradation products, including D-dimer, are found owing to the intense fibrinolytic activity stimulated by the presence of fibrin in the circulation. There is severe thrombocytopenia. The blood film may show fragmented red blood cells (schistocytes).
- Mild cases without bleeding: There is increased synthesis of coagulation factors and platelets. PT, APTT, and platelet counts are normal. fibrin degradation products are raised.
Definitive diagnosis depends on the result of:[11]
- Thrombocytopenia
- Prolongation of prothrombin time and activated partial thromboplastin time
- A low fibrinogen concentration
- Increased levels of fibrin degradation products
Condition Prothrombin time Partial thromboplastin time Bleeding time Platelet count Vitamin K deficiency or warfarin prolonged prolonged unaffected unaffected Disseminated intravascular coagulation prolonged prolonged prolonged decreased von Willebrand disease unaffected prolonged prolonged unaffected Haemophilia unaffected prolonged unaffected unaffected Aspirin unaffected unaffected prolonged unaffected Thrombocytopenia unaffected unaffected prolonged decreased Early Liver failure prolonged unaffected unaffected unaffected End-stage Liver failure prolonged prolonged prolonged decreased Uremia unaffected unaffected prolonged unaffected Congenital afibrinogenemia prolonged prolonged prolonged unaffected Factor V deficiency prolonged prolonged unaffected unaffected Factor X deficiency as seen in amyloid purpura prolonged prolonged unaffected unaffected Glanzmann's thrombasthenia unaffected unaffected prolonged unaffected Bernard-Soulier syndrome unaffected unaffected prolonged unaffected [12] Treatment
The only effective treatment is the reversal of the underlying cause. Anticoagulants are given exceedingly rarely when thrombus formation is likely to lead to imminent death (such as in coronary artery thrombosis or cerebrovascular thrombosis). Platelets may be transfused if counts are less than 5,000-10,000/mm3 and massive hemorrhage is occurring, and fresh frozen plasma may be administered in an attempt to replenish coagulation factors and anti-thrombotic factors, although these are only temporizing measures and may result in the increased development of thrombosis.
DIC results in lower fibrinogen levels (as it has all been converted to fibrin), and this can be tested for in the hospital lab. A more specific test is for "fibrin split products" (FSPs) or "fibrin degradation products" (FDPs) which are produced when fibrin undergoes degradation when blood clots are dissolved by fibrinolysis.
In some situations, infusion with antithrombin may be necessary.
Prognosis
Prognosis varies depending on the underlying disorder, and the extent of the intravascular thrombosis (clotting). The prognosis for those with DIC, regardless of cause, is often grim: Between 10% and 50% of patients will die.[13] DIC with sepsis (infection) has a significantly higher rate of death than DIC associated with trauma.[13]
One alternative interpretation of the acronym, "death is coming,"[14] refers to the lack of effective treatment options, and to the significant mortality associated with severe DIC.[15]
See also
References
- ^ Churchill Livingstone Pocket Medical Dictionary 14th Edition.
- ^ ISBN 0-443-07036-9 Davidson's Principles and Practice of Medicine 19th Edition. Churchill Livingstone. Page 200
- ^ a b c Robbins, Stanley L.; Cotran, Ramzi S.; Kumar, Vinay; Collins, Tucker (1999). Robbins' Pathologic Basis of Disease (6 ed.). Philadelphia: Saunders. ISBN 0-7216-7335-X.
- ^ a b Davidson, Stanley; Haslett, C. (2002). Davidson's Principles and Practice of Medicine (19 ed.). Edinburgh: Churchill Livingstone. ISBN 0-443-07036-9.
- ^ Kumar, Vinay; Abbas, Abul K.; Fausto, Nelson; & Mitchell, Richard N. (2007). Robbins Basic Pathology (8th ed.). Saunders Elsevier. pp. 469-471 ISBN 978-1-4160-2973-1
- ^ Rak J, Yu JL, Luyendyk J, Mackman N (2006). "Oncogenes, trousseau syndrome, and cancer-related changes in the coagulome of mice and humans". Cancer Res. 66 (22): 10643–6. doi:10.1158/0008-5472.CAN-06-2350. PMID 17108099. http://cancerres.aacrjournals.org/cgi/pmidlookup?view=long&pmid=17108099.
- ^ Grewal, P.K., Uchiyama, S., Ditto, D., Varki,N., Le, D.T., Nizet, V., and Marth, J.D. (2008). The Ashwell receptor mitigates the lethal coagulopathy of sepsis. Nat. Med., 14, 648-655.
- ^ a b c Clark, Michael; Kumar, Parveen J. (1998). Clinical Medicine: A Textbook for Medical Students and Doctors (4 ed.). Philadelphia: W.B. Saunders. ISBN 0-7020-2458-9.
- ^ Boyer EW, Shannon M (2005). "The serotonin syndrome". N. Engl. J. Med. 352 (11): 1112–20. doi:10.1056/NEJMra041867. PMID 15784664.
- ^ Oxford Handbook of Clinical Medicine 6th Edition. Page 650
- ^ ISBN 0-443-07036-9 Davidson's Principles and Practice of Medicine 19th Edition. Churchill Livingstone. Page 953.
- ^ Kaplan QBook - USMLE Step 1 - 5th edition - page 254
- ^ a b Becker, Joseph U and Charles R Wira. Disseminated intravascular coagulation at eMedicine, 10 September 2009
- ^ "Medical slang". RadsWiki. 25 July 2008. http://www.radswiki.net/main/index.php?title=Medical_slang. Retrieved 4 January 2010.
- ^ Norman K (2004). "Alternative treatments for disseminated intravascular coagulation.". Drug News Perspect 17 (4): 243–50. doi:10.1358/dnp.2004.17.4.829051. PMID 15334173.
Pathology: hematology · hematologic diseases of RBCs and megakaryocytes / MEP (D50-69,74, 280-287) Red
blood cells↑↓enzymopathy: G6PD · glycolysis (PK, TI, HK)
hemoglobinopathy: Thalassemia (alpha, beta, delta) · Sickle-cell disease/trait · HPFH
membrane: Hereditary spherocytosis (Minkowski-Chauffard syndrome) · Hereditary elliptocytosis (Southeast Asian ovalocytosis) · Hereditary stomatocytosisAcquiredHereditary: Fanconi anemia · Diamond–Blackfan anemia
Acquired: PRCA · Sideroblastic anemia · MyelophthisicOtherCoagulation/
coagulopathy↑primary: Antithrombin III deficiency · Protein C deficiency/Activated protein C resistance/Protein S deficiency/Factor V Leiden · Hyperprothrombinemia
acquired:Thrombocytosis (essential) · DIC (Congenital afibrinogenemia, Purpura fulminans) · autoimmune (Antiphospholipid)↓adhesion (Bernard–Soulier syndrome) · aggregation (Glanzmann's thrombasthenia) · platelet storage pool deficiency (Hermansky–Pudlak syndrome, Gray platelet syndrome)Clotting factorCategories:- Coagulopathies
- Intensive care medicine
- Vascular-related cutaneous conditions

Wikimedia Foundation. 2010.