- Sickle-cell disease
-
This article is about the disease itself. For the genetic transmission of sickle-cell disease and its carrier state, see sickle cell trait.
Sickle-cell disease Classification and external resources 
Normal and sickle-shaped red blood cellsICD-10 D57 ICD-9 282.6 OMIM 603903 DiseasesDB 12069 MedlinePlus 000527 eMedicine med/2126 oph/490 ped/2096 emerg/26 emerg/406 MeSH C15.378.071.141.150.150 GeneReviews Sickle-cell disease Sickle-cell disease (SCD), or sickle-cell anaemia (or anemia, SCA) or drepanocytosis, is an autosomal recessive genetic blood disorder with overdominance, characterized by red blood cells that assume an abnormal, rigid, sickle shape. Sickling decreases the cells' flexibility and results in a risk of various complications. The sickling occurs because of a mutation in the haemoglobin gene. Life expectancy is shortened, with studies reporting an average life expectancy of 42 in males and 48 in females.[1]
Sickle-cell disease, usually presenting in childhood, occurs more commonly in people (or their descendants) from parts of tropical and sub-tropical regions where malaria is or was common. One-third of all indigenous inhabitants of Sub-Saharan Africa carry the gene,[2] because in areas where malaria is common, there is a fitness benefit in carrying only a single sickle-cell gene (sickle cell trait). Those with only one of the two alleles of the sickle-cell disease, while not totally resistant, are more tolerant to the infection and thus show less severe symptoms when infected.[3]
The prevalence of the disease in the United States is approximately 1 in 5,000, mostly affecting Americans of Sub-Saharan African descent, according to the National Institutes of Health.[4] In the United States, about 1 out of 500 African-American children born will have sickle-cell anaemia.[5]
Sickle-cell anaemia is the name of a specific form of sickle-cell disease in which there is homozygosity for the mutation that causes HbS. Sickle-cell anaemia is also referred to as "HbSS", "SS disease", "haemoglobin S" or permutations thereof. In heterozygous people, who have only one sickle gene and one normal adult haemoglobin gene, it is referred to as "HbAS" or "sickle cell trait". Other, rarer forms of sickle-cell disease include sickle-haemoglobin C disease (HbSC), sickle beta-plus-thalassaemia (HbS/β+) and sickle beta-zero-thalassaemia (HbS/β0). These other forms of sickle-cell disease are compound heterozygous states in which the person has only one copy of the mutation that causes HbS and one copy of another abnormal haemoglobin allele.
The term disease is applied, because the inherited abnormality causes a pathological condition that can lead to death and severe complications. Not all inherited variants of haemoglobin are detrimental, a concept known as genetic polymorphism.
Contents
Signs and symptoms
Sickle-cell disease may lead to various acute and chronic complications, several of which have a high mortality rate.
Sickle cell crisis
The term "sickle cell crisis" is used to describe several independent acute conditions occurring in patients with sickle cell disease. Sickle cell disease results in anaemia and crisis that could be of many types including the vaso-occlusive crisis, aplastic crisis, sequestration crisis, haemolytic crisis and others. Most episodes of sickle cell crises last between five and seven days.[6]
Vaso-occlusive crisis
The vaso-occlusive crisis is caused by sickle-shaped red blood cells that obstruct capillaries and restrict blood flow to an organ, resulting in ischaemia, pain, necrosis and often organ damage. The frequency, severity, and duration of these crises vary considerably. Painful crises are treated with hydration, analgesics, and blood transfusion; pain management requires opioid administration at regular intervals until the crisis has settled. For milder crises, a subgroup of patients manage on NSAIDs (such as diclofenac or naproxen). For more severe crises, most patients require inpatient management for intravenous opioids; patient-controlled analgesia (PCA) devices are commonly used in this setting. Vaso-occlusive crisis involving organs such as the penis or lungs are considered an emergency and treated with red-blood cell transfusions. Diphenhydramine is sometimes effective for the itching associated with the opioid use. Incentive spirometry, a technique to encourage deep breathing to minimise the development of atelectasis, is recommended.[citation needed]
Splenic sequestration crisis
Because of its narrow vessels and function in clearing defective red blood cells, the spleen is frequently affected[citation needed]. It is usually infarcted before the end of childhood in individuals suffering from sickle-cell anaemia. This autosplenectomy increases the risk of infection from encapsulated organisms;[7][8] preventive antibiotics and vaccinations are recommended for those with such asplenia.
- Splenic sequestration crises: are acute, painful enlargements of the spleen. The sinusoids and gates would open at the same time resulting in sudden pooling of the blood into the spleen and circulatory defect leading to sudden hypovolaemia. The abdomen becomes bloated and very hard. Splenic sequestration crises is considered an emergency. If not treated, patients may die within 1–2 hours due to circulatory failure. Management is supportive, sometimes with blood transfusion. These crises are transient, they continue for 3–4 hours and may last for one day.
Aplastic crisis
Aplastic crises are acute worsenings of the patient's baseline anaemia, producing pallor, tachycardia, and fatigue. This crisis is triggered by parvovirus B19, which directly affects erythropoiesis (production of red blood cells) by invading the red cell precursors and multiplying in them and destroying them. Parvovirus infection nearly completely prevents red blood cell production for two to three days. In normal individuals, this is of little consequence, but the shortened red cell life of sickle-cell patients results in an abrupt, life-threatening situation. Reticulocyte counts drop dramatically during the disease (causing reticulocytopenia), and the rapid turnover of red cells leads to the drop in haemoglobin. This crisis takes 4 days to one week to disappear. Most patients can be managed supportively; some need blood transfusion.
Haemolytic crisis
Haemolytic crises are acute accelerated drops in haemoglobin level. The red blood cells break down at a faster rate. This is particularly common in patients with co-existent G6PD deficiency. Management is supportive, sometimes with blood transfusions.
Other
One of the earliest clinical manifestations is dactylitis, presenting as early as six months of age, and may occur in children with sickle trait.[9] The crisis can last up to a month.[10] Another recognised type of sickle crisis is the acute chest syndrome, a condition characterised by fever, chest pain, difficulty breathing, and pulmonary infiltrate on a chest X-ray. Given that pneumonia and sickling in the lung can both produce these symptoms, the patient is treated for both conditions.[citation needed] It can be triggered by painful crisis, respiratory infection, bone-marrow embolisation, or possibly by atelectasis, opiate administration, or surgery.
Complications
Sickle-cell anaemia can lead to various complications, including:
- Overwhelming post-(auto)splenectomy infection (OPSI), which is due to functional asplenia, caused by encapsulated organisms such as Streptococcus pneumoniae and Haemophilus influenzae. Daily penicillin prophylaxis is the most commonly used treatment during childhood, with some haematologists continuing treatment indefinitely. Patients benefit today from routine vaccination for H. influenzae, S. pneumoniae, and Neisseria meningitidis.
- Stroke, which can result from a progressive narrowing of blood vessels, preventing oxygen from reaching the brain. Cerebral infarction occurs in children and cerebral haemorrhage in adults.
- Silent stroke is a stroke that causes no immediate symptoms but is associated with damage to the brain. Silent stroke is probably five times as common as symptomatic stroke. Approximately 10–15% of children with sickle cell disease suffer strokes, with silent strokes predominating in the younger patients.[11][12]
- Cholelithiasis (gallstones) and cholecystitis, which may result from excessive bilirubin production and precipitation due to prolonged haemolysis.
- Avascular necrosis (aseptic bone necrosis) of the hip and other major joints, which may occur as a result of ischaemia.[13]
- Decreased immune reactions due to hyposplenism (malfunctioning of the spleen).[14]
- Priapism and infarction of the penis.[15]
- Osteomyelitis (bacterial bone infection); the most common cause of osteomyelitis in sickle cell disease is Salmonella (especially the non-typical serotypes Salmonella typhimurium, Salmonella enteritidis, Salmonella choleraesuis and Salmonella paratyphi B), followed by Staphylococcus aureus and Gram-negative enteric bacilli perhaps because intravascular sickling of the bowel leads to patchy ischaemic infarction.[16]
- Opioid tolerance, which can occur as a normal, physiologic response to the therapeutic use of opiates. Addiction to opiates occurs no more commonly among individuals with sickle-cell disease than among other individuals treated with opiates for other reasons.
- Acute papillary necrosis in the kidneys.
- Leg ulcers.[17]
- In eyes, background retinopathy, proliferative retinopathy, vitreous haemorrhages and retinal detachments, resulting in blindness.[18] Regular annual eye checks are recommended.
- During pregnancy, intrauterine growth retardation, spontaneous abortion, and pre-eclampsia.
- Chronic pain: Even in the absence of acute vaso-occlusive pain, many patients have chronic pain that is not reported.[19]
- Pulmonary hypertension (increased pressure on the pulmonary artery), leading to strain on the right ventricle and a risk of heart failure; typical symptoms are shortness of breath, decreased exercise tolerance and episodes of syncope.[20]
- Chronic renal failure due to Sickle cell nephropathy—manifests itself with hypertension (high blood pressure), proteinuria (protein loss in the urine), haematuria (loss of red blood cells in urine) and worsened anaemia. If it progresses to end-stage renal failure, it carries a poor prognosis.[21]
Heterozygotes
The heterozygous form (sickle cell trait) is almost always asymptomatic, and the only usual significant manifestation is the renal concentrating defect presenting with isosthenuria.
Pathophysiology
Sickle-cell anaemia is caused by a point mutation in the β-globin chain of haemoglobin, causing the hydrophilic amino acid glutamic acid to be replaced with the hydrophobic amino acid valine at the sixth position. The β-globin gene is found on chromosome 16, (Robbin's Pathology). The association of two wild-type α-globin subunits with two mutant β-globin subunits forms haemoglobin S (HbS). Under low-oxygen conditions (being at high altitude, for example), the absence of a polar amino acid at position six of the β-globin chain promotes the non-covalent polymerisation (aggregation) of haemoglobin, which distorts red blood cells into a sickle shape and decreases their elasticity.
The loss of red blood cell elasticity is central to the pathophysiology of sickle-cell disease. Normal red blood cells are quite elastic, which allows the cells to deform to pass through capillaries. In sickle-cell disease, low-oxygen tension promotes red blood cell sickling and repeated episodes of sickling damage the cell membrane and decrease the cell's elasticity. These cells fail to return to normal shape when normal oxygen tension is restored. As a consequence, these rigid blood cells are unable to deform as they pass through narrow capillaries, leading to vessel occlusion and ischaemia.
The actual anaemia of the illness is caused by haemolysis, the destruction of the red cells inside the spleen, because of their misshape. Although the bone marrow attempts to compensate by creating new red cells, it does not match the rate of destruction.[22] Healthy red blood cells typically live 90–120 days, but sickle cells only survive 10–20 days.[23]
Normally, humans have Haemoglobin A, which consists of two alpha and two beta chains, Haemoglobin A2, which consists of two alpha and two delta chains and Haemoglobin F, consisting of two alpha and two gamma chains in their bodies. Of these, Haemoglobin A makes up around 96-97% of the normal haemoglobin in humans.
Genetics
Sickle-cell gene mutation probably arose spontaneously in different geographic areas, as suggested by restriction endonuclease analysis. These variants are known as Cameroon, Senegal, Benin, Bantu and Saudi-Asian. Their clinical importance springs from the fact that some of them are associated with higher HbF levels, e.g., Senegal and Saudi-Asian variants, and tend to have milder disease.[24]
In people heterozygous for HgbS (carriers of sickling haemoglobin), the polymerisation problems are minor, because the normal allele is able to produce over 50% of the haemoglobin. In people homozygous for HgbS, the presence of long-chain polymers of HbS distort the shape of the red blood cell from a smooth doughnut-like shape to ragged and full of spikes, making it fragile and susceptible to breaking within capillaries. Carriers have symptoms only if they are deprived of oxygen (for example, while climbing a mountain) or while severely dehydrated. Under normal circumstances, these painful crises occur about 0.8 times per year per patient.[citation needed] The sickle-cell disease occurs when the seventh amino acid (if the initial methionine is counted), glutamic acid, is replaced by valine to change its structure and function. Valine is hydrophobic, causing the haemoglobin to collapse in on itself occasionally. The structure is not changed otherwise. When enough haemoglobin collapses in on itself the red blood cells become sickle-shaped.
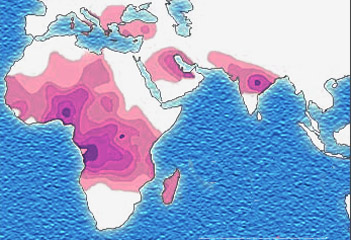
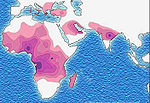 Distribution of the sickle-cell trait shown in pink and purple
Distribution of the sickle-cell trait shown in pink and purple
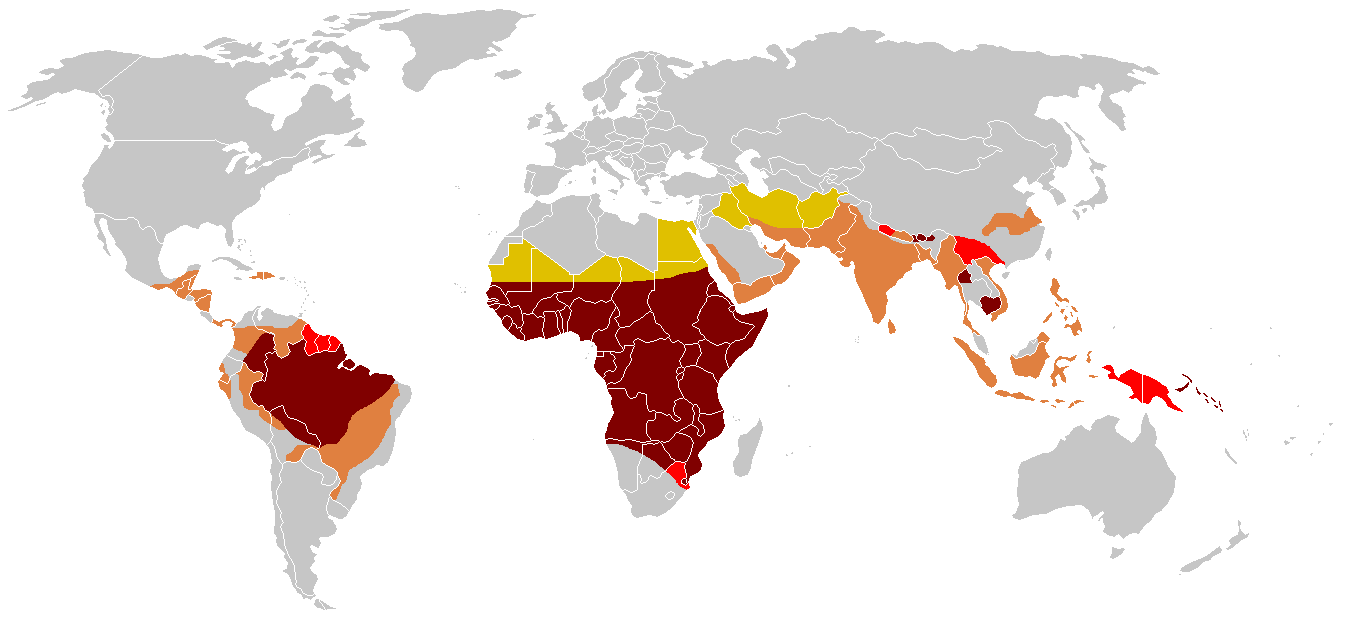
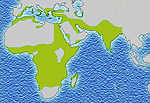 Historical distribution of malaria (no longer endemic in Europe) shown in green
Historical distribution of malaria (no longer endemic in Europe) shown in green Modern distribution of malaria
Modern distribution of malariaThe gene defect is a known mutation of a single nucleotide (see single-nucleotide polymorphism - SNP) (A to T) of the β-globin gene, which results in glutamic acid being substituted by valine at position 6. Haemoglobin S with this mutation is referred to as HbS, as opposed to the normal adult HbA. The genetic disorder is due to the mutation of a single nucleotide, from a GAG to GTG codon mutation, becoming a GUG codon by transcription. This is normally a benign mutation, causing no apparent effects on the secondary, tertiary, or quaternary structure of haemoglobin in conditions of normal oxygen concentration. What it does allow for, under conditions of low oxygen concentration, is the polymerization of the HbS itself. The deoxy form of haemoglobin exposes a hydrophobic patch on the protein between the E and F helices. The hydrophobic residues of the valine at position 6 of the beta chain in haemoglobin are able to associate with the hydrophobic patch, causing haemoglobin S molecules to aggregate and form fibrous precipitates.
The allele responsible for sickle-cell anaemia is autosomal recessive and can be found on the short arm of chromosome 11. A person that receives the defective gene from both father and mother develops the disease; a person that receives one defective and one healthy allele remains healthy, but can pass on the disease and is known as a carrier. If two parents who are carriers have a child, there is a 1-in-4 chance of their child developing the disease and a 1-in-2 chance of their child's being just a carrier. Since the gene is incompletely recessive, carriers can produce a few sickled red blood cells, not enough to cause symptoms, but enough to give resistance to malaria. Because of this, heterozygotes have a higher fitness than either of the homozygotes. This is known as heterozygote advantage.
Due to the adaptive advantage of the heterozygote, the disease is still prevalent, especially among people with recent ancestry in malaria-stricken areas, such as Africa, the Mediterranean, India and the Middle East.[25] Malaria was historically endemic to southern Europe, but it was declared eradicated in the mid-20th century, with the exception of rare sporadic cases.[26]
The malaria parasite has a complex life cycle and spends part of it in red blood cells. In a carrier, the presence of the malaria parasite causes the red blood cells with defective haemoglobin to rupture prematurely, making the plasmodium unable to reproduce. Further, the polymerization of Hb affects the ability of the parasite to digest Hb in the first place. Therefore, in areas where malaria is a problem, people's chances of survival actually increase if they carry sickle-cell trait (selection for the heterozygote).
In the USA, where there is no endemic malaria, the prevalence of sickle-cell anaemia among blacks is lower (about 0.25%) than in West Africa (about 4.0%) and is falling. Without endemic malaria, the sickle cell mutation is purely disadvantageous and will tend to be selected out of the affected population. However, the so-called African American community of the USA is known to be the result of significant admixture between several African and non-African ethnic groups, and also represents the descendants of survivors of the slavery and the slave trade. Thus, a lower degree of endogamy and, particularly, abnormally high health-selective pressure through slavery may be the most plausible explanations for the lower prevalence of sickle-cell anaemia (and, possibly, other genetic diseases) among Afro-Americans compared to Sub-Saharan African people. Another factor limiting the spread of sickle-cell genes in North America is the absence of cultural proclivities to polygamy, which allows affected males to continue to seek unaffected children with multiple partners.[27]
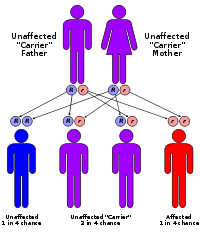 Sickle-cell disease is inherited in the autosomal recessive pattern.
Sickle-cell disease is inherited in the autosomal recessive pattern.Inheritance
Sickle-cell conditions are inherited from parents in much the same way as blood type, hair colour and texture, eye colour, and other physical traits. The types of haemoglobin a person makes in the red blood cells depend on what haemoglobin genes are inherited from his parents. If one parent has sickle-cell anaemia (SS) and the other has sickle-cell trait (AS), there is a 50% chance of a child's having sickle-cell disease (SS) and a 50% chance of a child's having sickle-cell trait (AS). When both parents have sickle-cell trait (AS), a child has a 25% chance (1 of 4) of sickle-cell disease (SS), as shown in the diagram.
Epidemiology
The highest frequency of sickle cell disease is found in tropical regions, particularly sub-Saharan Africa, India and the Middle-East.[28] Migration of substantial populations from these high prevalence areas to low prevalence countries in Europe has dramatically increased in recent decades and in some European countries sickle cell disease has now overtaken more familiar genetic conditions such as haemophilia and cystic fibrosis.[29]
Africa
Three quarters of sickle-cell cases occur in Africa. A recent WHO report estimated that around 2% of newborns in Nigeria were affected by sickle cell anaemia, giving a total of 150,000 affected children born every year in Nigeria alone. The carrier frequency ranges between 10% and 40% across equatorial Africa, decreasing to 1–2% on the north African coast and <1% in South Africa.[30]
Europe
France
In Europe, the highest prevalence of the disease has been observed in France. As a result of population growth in African-Caribbean regions of overseas France, and now immigration essentially from North and sub-Saharan Africa to mainland France, sickle cell disease has become a major health problem in France.[31] SCD has become the most common genetic disease in this country, with an overall birth prevalence of 1/2,415 in mainland France, ahead of phenylketonuria (1/10,862), congenital hypothyroidism (1/3,132), congenital adrenal hyperplasia (1/19,008) and cystic fibrosis (1/5,014) for the same reference period. In 2007, 28.45% of all newborns in mainland France had at least one parent originated from a region defined "at risk" (mainly Africa and Overseas departments and territories of France) and were screened for SCD. The Paris metropolitan district (Île-de-France) is the region that accounts for the largest number of people at presumably higher risk of SCD. Indeed, nearly 56% of all newborns in this area in 2007 had at least one parent originated from a region defined as "at-risk" and were screened for SCD. The second largest number of at-risk is in Provence-Alpes-Côte d'Azur at nearly 42% and the lowest number is in Brittany at 4.40%.[32]
United Kingdom
In United Kingdom, more than 200 babies are born annually with SCD.
Middle East
About 6,000 children are born annually with SCD, at least 50% of these in Saudi Arabia, especially in Qatif City.
India
Sickle cell disease is prevalent in many parts of India, where the prevalence has ranged from 9.4 to 22.2% in endemic areas.[33]
Diagnosis
In HbSS, the full blood count reveals haemoglobin levels in the range of 6–8 g/dL with a high reticulocyte count (as the bone marrow compensates for the destruction of sickle cells by producing more red blood cells). In other forms of sickle-cell disease, Hb levels tend to be higher. A blood film may show features of hyposplenism (target cells and Howell-Jolly bodies).
Sickling of the red blood cells, on a blood film, can be induced by the addition of sodium metabisulfite. The presence of sickle haemoglobin can also be demonstrated with the "sickle solubility test". A mixture of haemoglobin S (Hb S) in a reducing solution (such as sodium dithionite) gives a turbid appearance, whereas normal Hb gives a clear solution.
Abnormal haemoglobin forms can be detected on haemoglobin electrophoresis, a form of gel electrophoresis on which the various types of haemoglobin move at varying speeds. Sickle-cell haemoglobin (HgbS) and haemoglobin C with sickling (HgbSC)—the two most common forms—can be identified from there. The diagnosis can be confirmed with high-performance liquid chromatography (HPLC). Genetic testing is rarely performed, as other investigations are highly specific for HbS and HbC.[34]
An acute sickle-cell crisis is often precipitated by infection. Therefore, a urinalysis to detect an occult urinary tract infection, and chest X-ray to look for occult pneumonia should be routinely performed.[35]
People who are known carriers of the disease often undergo genetic counseling before they have a child. A test to see if an unborn child has the disease takes either a blood sample from the fetus or a sample of amniotic fluid. Since taking a blood sample from a fetus has greater risks, the latter test is usually used.
After the mutation responsible for this disease was discovered in 1979, the U.S. Air Force required black applicants to test for the mutation. It dismissed 143 applicants because they were carriers, even though none of them had the condition. It eventually withdrew the requirement, but only after a trainee filed a lawsuit.[36]
Management
Folic acid and penicillin
Children born with sickle-cell disease will undergo close observation by the paediatrician and will require management by a haematologist to assure they remain healthy. These patients will take a 1 mg dose of folic acid daily for life. From birth to five years of age, they will also have to take penicillin daily due to the immature immune system that makes them more prone to early childhood illnesses.
Malaria chemoprophylaxis
The protective effect of sickle cell trait does not apply to people with sickle cell disease; in fact, they are uniquely vulnerable to malaria, since the most common cause of painful crises in malarial countries is infection with malaria. It has therefore been recommended that people with sickle cell disease living in malarial countries should receive anti-malarial chemoprophylaxis for life.[37]
Vaso-occlusive crisis
Most people with sickle-cell disease have intensely painful episodes called vaso-occlusive crises. The frequency, severity, and duration of these crises, however, vary tremendously. Painful crises are treated symptomatically with analgesics; pain management requires opioid administration at regular intervals until the crisis has settled. For milder crises, a subgroup of patients manage on NSAIDs (such as diclofenac or naproxen). For more severe crises, most patients require inpatient management for intravenous opioids; patient-controlled analgesia (PCA) devices are commonly used in this setting. Diphenhydramine is also an effective agent that is frequently prescribed by doctors in order to help control any itching associated with the use of opioids.
Acute chest crisis
Management is similar to vaso-occlusive crisis, with the addition of antibiotics (usually a quinolone or macrolide, since wall-deficient ["atypical"] bacteria are thought to contribute to the syndrome),[38] oxygen supplementation for hypoxia, and close observation. Should the pulmonary infiltrate worsen or the oxygen requirements increase, simple blood transfusion or exchange transfusion is indicated. The latter involves the exchange of a significant portion of the patients red cell mass for normal red cells, which decreases the percent of haemoglobin S in the patient's blood.
Hydroxyurea
The first approved drug for the causative treatment of sickle-cell anaemia, hydroxyurea, was shown to decrease the number and severity of attacks in a study in 1995 (Charache et al.)[39] and shown to possibly increase survival time in a study in 2003 (Steinberg et al.).[40] This is achieved, in part, by reactivating fetal haemoglobin production in place of the haemoglobin S that causes sickle-cell anaemia. Hydroxyurea had previously been used as a chemotherapy agent, and there is some concern that long-term use may be harmful, but this risk has been shown to be either absent or very small and it is likely that the benefits outweigh the risks.[41]
Transfusion therapy
Blood transfusions are often used in the management of sickle cell disease in acute cases and to prevent complications by decreasing the the number of red blood cells (RBC) that can sickle by adding normal red blood cells.[42] In children prophylactic chronic red blood cell (RBC) transfusion therapy has been shown to be efficacious to a certain extent in reducing the risk of first stroke or silent stroke when transcranial Doppler (TCD) ultrasonography shows abnormal increased cerebral blood flow velocities. In those who have sustained a prior stoke event it also reduces the risk of recurrent stroke and additional silent strokes.[43][44]
Bone marrow transplants
Bone marrow transplants have proven to be effective in children.[45]
History
This collection of clinical findings was unknown until the explanation of the sickle cells in 1910 by the Chicago cardiologist and professor of medicine James B. Herrick (1861–1954), whose intern Ernest Edward Irons (1877–1959) found "peculiar elongated and sickle-shaped" cells in the blood of Walter Clement Noel, a 20-year-old first-year dental student from Grenada, after Noel was admitted to the Chicago Presbyterian Hospital in December 1904 suffering from anaemia.[46]
Noel was readmitted several times over the next three years for "muscular rheumatism" and "bilious attacks". Noel completed his studies and returned to the capital of Grenada (St. George's) to practice dentistry. He died of pneumonia in 1916 and is buried in the Catholic cemetery at Sauteurs in the north of Grenada.[47] Herrick's published account included illustrations, but the earliest available slide showing sickle cells is that of a 1918 autopsy from a soldier with sickle trait, initially reviewed only 92 years later.[48]
The disease was named "sickle-cell anaemia" by Verne Mason in 1922, then a medical resident at Johns Hopkins Hospital.[49] However, some elements of the disease had been recognized earlier: A paper in the Southern Journal of Medical Pharmacology in 1846 described the absence of a spleen in the autopsy of a runaway slave. The African medical literature reported this condition in the 1870s, when it was known locally as ogbanjes ("children who come and go") because of the very high infant mortality rate caused by this condition. A history of the condition tracked reports back to 1670 in one Ghanaian family.[50] Also, the practice of using tar soap to cover blemishes caused by sickle-cell sores was prevalent in the black community.[citation needed]
Linus Pauling and colleagues were the first, in 1949, to demonstrate that sickle-cell disease occurs as a result of an abnormality in the haemoglobin molecule. This was the first time a genetic disease was linked to a mutation of a specific protein, a milestone in the history of molecular biology, and it was published in their paper "Sickle Cell Anemia, a Molecular Disease".
See also
- Haematopoietic ulcer
- African admixture in Europe
References
- ^ Platt OS, Brambilla DJ, Rosse WF, et al. (June 1994). "Mortality in sickle cell disease. Life expectancy and risk factors for early death". N. Engl. J. Med. 330 (23): 1639–44. doi:10.1056/NEJM199406093302303. ISSN 0028-4793. PMID 7993409. http://content.nejm.org/cgi/content/full/330/23/1639.
- ^ Sicklecell.md
- ^ Wellems TE, Hayton K, Fairhurst RM (September 2009). "The impact of malaria parasitism: from corpuscles to communities". J. Clin. Invest. 119 (9): 2496–505. doi:10.1172/JCI38307. PMC 2735907. PMID 19729847. http://www.jci.org/articles/view/38307.
- ^ National Heart, Lung and Blood Institute. "Sickle cell anemia, key points". http://www.nhlbi.nih.gov/health/dci/Diseases/Sca/SCA_Summary.html. Retrieved 2010-11-27.
- ^ "September is Sickle Cell Awareness Month". CDC. http://www.cdc.gov/Features/SickleCellAwareness/. Retrieved 6 February 2011.
- ^ "BestBets: How long should an average sickle cell crisis last?". http://www.bestbets.org/bets/bet.php?id=1189. Retrieved 2010-11-27.
- ^ Pearson H (Aug 1977). "Sickle cell anemia and severe infections due to encapsulated bacteria" (Free full text). J Infect Dis 136 Suppl: S25–30. ISSN 0022-1899. PMID 330779. http://www.nlm.nih.gov/medlineplus/meningitis.html.
- ^ Wong W, Powars D, Chan L, Hiti A, Johnson C, Overturf G (Mar 1992). "Polysaccharide encapsulated bacterial infection in sickle cell anaemia: a thirty year epidemiologic experience". Am J Hematol 39 (3): 176–82. doi:10.1002/ajh.2830390305. PMID 1546714.
- ^ Jadavji T, Prober CG (April 1985). "Dactylitis in a child with sickle cell trait". Can Med Assoc J 132 (7): 814–5. ISSN 0008-4409. PMC 1345873. PMID 3978504. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1345873.
- ^ http://www.ejbjs.org/cgi/content/abstract/58/8/1161
- ^ Adams RJ, Ohene-Frempong K, Wang W (2001). "Sickle cell and the brain". Hematology Am Soc Hematol Educ Program 2001 (1): 31–46. doi:10.1182/asheducation-2001.1.31. PMID 11722977. http://asheducationbook.hematologylibrary.org/cgi/content/full/2001/1/31.
- ^ Adams RJ (November 2007). "Big strokes in small persons". Arch. Neurol. 64 (11): 1567–74. doi:10.1001/archneur.64.11.1567. PMID 17998439. http://archneur.ama-assn.org/cgi/content/full/64/11/1567.
- ^ Martí-Carvajal, A; Dunlop, R; Agreda-Perez, L (Oct 2004). Martí-Carvajal, Arturo J. ed. "Treatment for avascular necrosis of bone in people with sickle cell disease". Cochrane database of systematic reviews (Online) (4): CD004344. doi:10.1002/14651858.CD004344.pub2. PMID 15495103.
- ^ Kenny MW, George AJ, Stuart J (July 1980). "Platelet hyperactivity in sickle-cell disease: a consequence of hyposplenism". Journal of Clinical Pathology 33 (7): 622–5. doi:10.1136/jcp.33.7.622. PMC 1146172. PMID 7430367. http://jcp.bmj.com/cgi/pmidlookup?view=long&pmid=7430367. Retrieved 2011-03-23.
- ^ Chrouser KL, Ajiboye OB, Oyetunji TA, Chang DC (April 2011). "Priapism in the United States: the changing role of sickle cell disease". American Journal of Surgery 201 (4): 468–74. doi:10.1016/j.amjsurg.2010.03.017. PMID 21421100.
- ^ Almeida A, Roberts I (May 2005). "Bone involvement in sickle cell disease". Br. J. Haematol. 129 (4): 482–90. doi:10.1111/j.1365-2141.2005.05476.x. PMID 15877730. http://www3.interscience.wiley.com/cgi-bin/fulltext/118642709/HTMLSTART.
- ^ Rudge FW (1991). "Hyperbaric Oxygen Therapy in the Treatment of Sickle Cell Leg Ulcers". J. Hyperbaric Med 6 (1): 1–4. http://archive.rubicon-foundation.org/4442. Retrieved 2011-03-23.
- ^ Elagouz M, Jyothi S, Gupta B, Sivaprasad S (July 2010). "Sickle cell disease and the eye: old and new concepts". Survey of Ophthalmology 55 (4): 359–77. doi:10.1016/j.survophthal.2009.11.004. PMID 20452638. http://linkinghub.elsevier.com/retrieve/pii/S0039-6257(09)00307-5. Retrieved 2011-03-23.
- ^ Smith WR, Penberthy LT, Bovbjerg VE, et al. (Jan 2008). "Daily assessment of pain in adults with sickle cell disease". Ann. Intern. Med. 148 (2): 94–101. ISSN 0003-4819. PMID 18195334.
- ^ Gladwin MT, Sachdev V, Jison ML, et al. (February 2004). "Pulmonary hypertension as a risk factor for death in patients with sickle cell disease". N. Engl. J. Med. 350 (9): 886–95. doi:10.1056/NEJMoa035477. PMID 14985486. http://www.nejm.org/doi/full/10.1056/NEJMoa035477#t=article.
- ^ Powars DR, Elliott-Mills DD, Chan L, et al. (Oct 1991). "Chronic renal failure in sickle cell disease: risk factors, clinical course, and mortality". Ann. Intern. Med. 115 (8): 614–20. ISSN 0003-4819. PMID 1892333.
- ^ "How Does Sickle Cell Cause Disease?". http://sickle.bwh.harvard.edu/scd_background.html. Retrieved 2010-11-27.
- ^ "Sickle Cell Anemia: eMedicine Emergency Medicine". http://emedicine.medscape.com/article/778971-overview. Retrieved 2010-11-27.
- ^ Green NS, Fabry ME, Kaptue-Noche L, Nagel RL (Oct 1993). "Senegal haplotype is associated with higher HbF than Benin and Cameroon haplotypes in African children with sickle cell anemia". Am. J. Hematol. 44 (2): 145–6. doi:10.1002/ajh.2830440214. ISSN 0361-8609. PMID 7505527.
- ^ Kwiatkowski DP (Aug 2005). "How Malaria Has Affected the Human Genome and What Human Genetics Can Teach Us about Malaria". Am. J. Hum. Genet. 77 (2): 171–92. doi:10.1086/432519. ISSN 0002-9297. PMC 1224522. PMID 16001361. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1224522. Full text at PMC: 1224522
- ^ Ponçon N, Toty C, L'Ambert G, et al. (2007). "Biology and dynamics of potential malaria vectors in Southern France". Malar. J. 6: 18. doi:10.1186/1475-2875-6-18. PMC 1808464. PMID 17313664. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1808464.
- ^ Lesi FE, Bassey EE (July 1972). "Family study in sickle cell disease in Nigeria". J Biosoc Sci 4 (3): 307–13. doi:10.1017/S0021932000008622. PMID 5041262.
- ^ Weatherall DJ, Clegg JB (2001). "Inherited haemoglobin disorders: an increasing global health problem". Bull. World Health Organ. 79 (8): 704–12. PMC 2566499. PMID 11545326. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2566499.
- ^ Roberts I, de Montalembert M (July 2007). "Sickle cell disease as a paradigm of immigration hematology: new challenges for hematologists in Europe". Haematologica 92 (7): 865–71. doi:10.3324/haematol.11474. PMID 17606434. http://www.haematologica.org/cgi/content/full/92/7/865.
- ^ WHO. "Sickle-cell anaemia - Report by the Secretariat" (PDF). http://apps.who.int/gb/ebwha/pdf_files/WHA59/A59_9-en.pdf. Retrieved 2010-11-27.
- ^ Bardakdjian J, Wajcman H (September 2004). "[Epidemiology of sickle cell anemia]" (in French). Rev Prat 54 (14): 1531–3. PMID 15558961.
- ^ Bardakdjian-Michau J, Bahuau M, Hurtrel D, et al. (January 2009). "Neonatal screening for sickle cell disease in France". J. Clin. Pathol. 62 (1): 31–3. doi:10.1136/jcp.2008.058867. PMID 19103855.
- ^ Awasthy N, Aggarwal KC, Goyal PC, Prasad MS, Saluja S, Sharma M (2008). "Sickle cell disease: Experience of a tertiary care center in a nonendemic area". Annals of Tropical Medicine and Public Health 1 (1): 1–4. doi:10.4103/1755-6783.43069.
- ^ Clarke GM, Higgins TN (August 2000). "Laboratory investigation of hemoglobinopathies and thalassemias: review and update". Clin. Chem. 46 (8 Pt 2): 1284–90. PMID 10926923. http://www.clinchem.org/cgi/content/full/46/8/1284.
- ^ "BestBets: Does routine urinalysis and chest radiography detect occult bacterial infection in sickle cell patients presenting to the accident and emergency department with painful crisis?". http://www.bestbets.org/bets/bet.php?id=1102. Retrieved 2010-11-27.
- ^ Anonymous (4 January 1981). "Air force academy sued over sickle cell policy". New York Times. http://query.nytimes.com/gst/fullpage.html?sec=health&res=9807EFD7163BF937A35752C0A967948260. Retrieved 21 December 2008.
- ^ Oniyangi O, Omari AA (2006). Oniyangi, Oluseyi. ed. "Malaria chemoprophylaxis in sickle cell disease". Cochrane database of systematic reviews 13 (4): CD003489. doi:10.1002/14651858.CD003489.pub2. PMID 17054173.
- ^ Aldrich TK, Nagel RL. (1998). "Pulmonary Complications of Sickle Cell Disease.". In Reynolds HY, Bone RC, Dantzker DR, George RB, Matthay RA. Pulmonary and Critical Care Medicine (6th ed.). St. Louis: Mosby. pp. 1–10. ISBN 0-8151-1371-4.
- ^ Charache S, Terrin ML, Moore RD, et al. (May 1995). "Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia". N. Engl. J. Med. 332 (20): 1317–22. doi:10.1056/NEJM199505183322001. ISSN 0028-4793. PMID 7715639.
- ^ Steinberg MH, Barton F, Castro O, et al. (April 2003). "Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment". JAMA 289 (13): 1645–51. doi:10.1001/jama.289.13.1645. PMID 12672732. http://jama.ama-assn.org/cgi/content/full/289/13/1645.
- ^ Platt OS (Mar 2008). "Hydroxyurea for the treatment of sickle cell anemia". N. Engl. J. Med. 358 (13): 1362–9. doi:10.1056/NEJMct0708272. PMID 18367739.
- ^ Drasar E, Igbineweka N, Vasavda N, et al. Blood transfusion usage among adults with sickle cell disease - a single institution experience over ten years. Br J Haematol. 2011 Mar;152(6):766-70. doi: 10.1111/j.1365-2141.2010.08451.x. Epub 2011 Jan 31. PMID 21275951
- ^ Gyang E, Yeom K, Hoppe C, et al. Effect of chronic red cell transfusion therapy on vasculopathies and silent infarcts in patients with sickle cell disease. Am J Hematol. 2011 Jan;86(1):104-6. PMID 21117059
- ^ Mirre E, Brousse V, Berteloot L, et al. Feasibility and efficacy of chronic transfusion for stroke prevention in children with sickle cell disease Eur J Haematol. 2010 Mar;84(3):259-65. Epub 2009 Nov 12. PMID 19912310
- ^ Walters MC, Patience M, Leisenring W, et al. (August 1996). "Bone marrow transplantation for sickle cell disease". N. Engl. J. Med. 335 (6): 369–76. doi:10.1056/NEJM199608083350601. PMID 8663884. http://www.nejm.org/doi/full/10.1056/NEJM199608083350601.
- ^ Herrick JB (1910). "Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia". Arch. Intern. Med. 6: 517–521. doi:10.1001/archinte.1910.00050330050003. http://archinte.ama-assn.org/cgi/content/summary/VI/5/517.; reprinted as Herrick JB (2001). "Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. 1910". The Yale journal of biology and medicine 74 (3): 179–84. PMC 2588723. PMID 11501714. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2588723.
- ^ Savitt TL, Goldberg MF (Jan 1989). "Herrick's 1910 case report of sickle cell anemia. The rest of the story". JAMA 261 (2): 266–71. doi:10.1001/jama.261.2.266. PMID 2642320.
- ^ "From 1918 Autopsy, A First Glimpse of Sickle Cell — and a Warning". http://www.wired.com/wiredscience/2010/11/from-a-1918-autopsy-a-first-glimpse-of-sickle-cell-%E2%80%94%C2%A0and-a-warning. Retrieved 2010-11-28.
- ^ Mason VR (1922). "Sickle cell anemia". JAMA 79 (14): 1318–1320. doi:10.1001/jama.254.14.1955. PMID 3900438.
- ^ Konotey-Ahulu FID. Effect of environment on sickle cell disease in West Africa: epidemiologic and clinical considerations. In: Sickle Cell Disease, Diagnosis, Management, Education and Research. Abramson H, Bertles JF, Wethers DL, eds. CV Mosby Co, St. Louis. 1973; 20; cited in Desai, D. V.; Hiren Dhanani (2004). "Sickle Cell Disease: History And Origin". The Internet Journal of Haematology 1 (2). ISSN 1540-2649. http://www.ispub.com/ostia/index.php?xmlFilePath=journals/ijhe/vol1n2/sickle.xml.
Further reading
- Brown, Robert T., ed (2006). Comprehensive handbook of childhood cancer and sickle cell disease: a biopsychosocial approach. Oxford University Press. ISBN 9780195169850. http://books.google.com/books?id=O2sq8EeKTdwC.
- Hill, Shirley A. (2003). Managing Sickle Cell Disease in Low-Income Families. Temple University Press. ISBN 9781592131952. http://books.google.com/books?id=YG0Oi9mAaoQC.
- Serjeant, Graham R. & Beryl E. (2001). Sickle Cell Disease. Oxford University Press. ISBN 9780192630360. http://books.google.com/books?id=SetrAAAAMAAJ.
- Tapper, Melbourne (1999). In the blood: sickle cell anemia and the politics of race. University of Pennsylvania Press. ISBN 9780812234718. http://books.google.com/books?id=YzM0oARlXvIC.
External links
Categories:- Autosomal recessive disorders
- Chronic pain syndromes
- Hereditary hemolytic anemias
- Health in Africa
- Hematopathology
- Disorders of globin and globulin proteins


Wikimedia Foundation. 2010.