- Phenylketonuria
-
Phenylketonuria Classification and external resources ICD-10 E70.0 ICD-9 270.1 OMIM 261600 261630 DiseasesDB 9987 MedlinePlus 001166 eMedicine ped/1787 derm/712 MeSH D010661 Phenylketonuria (PKU) is an autosomal recessive metabolic genetic disorder characterized by a mutation in the gene for the hepatic enzyme phenylalanine hydroxylase (PAH), rendering it nonfunctional.[1]:541 This enzyme is necessary to metabolize the amino acid phenylalanine (Phe) to the amino acid tyrosine. When PAH activity is reduced, phenylalanine accumulates and is converted into phenylpyruvate (also known as phenylketone), which is detected in the urine.[2]
Since its discovery, there have been many advances in its treatment. It can now be successfully managed by the patient under ongoing medical supervision to avoid the more serious side effects. If, however, the condition is left untreated, it can cause problems with brain development, leading to progressive mental retardation, brain damage, and seizures. Early cases of PKU were treated with a low-phenylalanine diet. More recent research has now shown that diet alone may not be enough to prevent the negative effects of elevated phenylalanine levels. Optimal treatment involves maintaining blood Phe levels in a safe range while monitoring diet and cognitive development. There is no cure for PKU, but patients who are diagnosed early and maintain a strict diet can have a normal life span with normal mental development.
Contents
History
Phenylketonuria was discovered by the Norwegian physician Ivar Asbjørn Følling in 1934[3] when he noticed hyperphenylalaninemia (HPA) was associated with mental retardation. In Norway, this disorder is known as Følling's disease, named after its discoverer.[4] Dr. Følling was one of the first physicians to apply detailed chemical analysis to the study of disease. His careful analysis of the urine of two affected siblings led him to request many physicians near Oslo to test the urine of other affected patients. This led to the discovery of the same substance he had found in eight other patients. He conducted tests and found reactions that gave rise to benzaldehyde and benzoic acid, which led him to conclude that the compound contained a benzene ring. Further testing showed the melting point to be the same as phenylpyruvic acid, which indicated that the substance was in the urine. His careful science inspired many to pursue similar meticulous and painstaking research with other disorders.
Screening and presentation
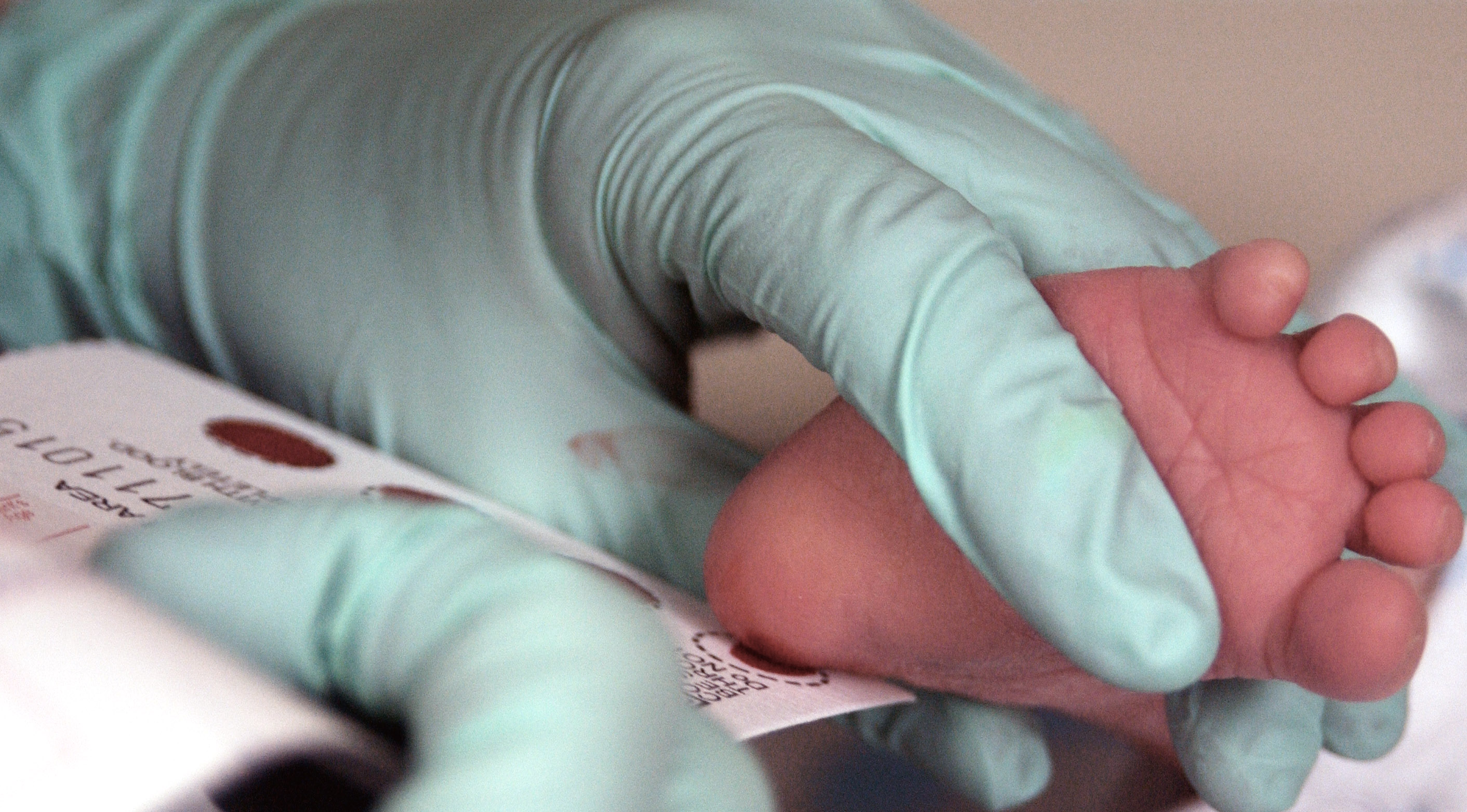
 Blood is taken from a two-week old infant to test for phenylketonuria
Blood is taken from a two-week old infant to test for phenylketonuria
PKU is commonly included in the newborn screening panel of most countries, with varied detection techniques. Most babies in developed countries are screened for PKU soon after birth.[5] Screening for PKU is done with bacterial inhibition assay (Guthrie test), immunoassays using fluorometric or photometric detection, or amino acid measurement using tandem mass spectrometry (MS/MS). Measurements done using MS/MS determine the concentration of Phe and the ratio of Phe to tyrosine, both of which will be elevated in PKU.[6]
If a child is not screened during the routine newborn screening test (typically performed 2 - 7 days after birth, using samples drawn by neonatal heel prick), the disease may present clinically with seizures, albinism (excessively fair hair and skin), and a "musty odor" to the baby's sweat and urine (due to phenylacetate, one of the ketones produced). In most cases, a repeat test should be done at approximately two weeks of age to verify the initial test and uncover any phenylketonuria that was initially missed.
Untreated children are normal at birth, but fail to attain early developmental milestones, develop microcephaly, and demonstrate progressive impairment of cerebral function. Hyperactivity, EEG abnormalities and seizures, and severe learning disabilities are major clinical problems later in life. A "musty or mousy" odor of skin, hair, sweat and urine (due to phenylacetate accumulation); and a tendency to hypopigmentation and eczema are also observed.
In contrast, affected children who are detected and treated are less likely to develop neurological problems or have seizures and mental retardation, though such clinical disorders are still possible.
Pathophysiology
Classical PKU
Classical PKU is caused by a mutated gene for the enzyme phenylalanine hydroxylase (PAH), which converts the amino acid phenylalanine to other essential compounds in the body. Other non-PAH mutations can also cause PKU. This is an example of genetic heterogeneity. The PAH gene is located on chromosome 12 in the bands 12q22-q24.1. More than 400 disease-causing mutations have been found in the PAH gene. PAH deficiency causes a spectrum of disorders, including classic phenylketonuria (PKU) and hyperphenylalaninemia (a less severe accumulation of phenylalanine).[7]
PKU is known to be an autosomal recessive genetic disorder. This means both parents must have at least one mutated allele of the PAH gene. The child must inherit both mutated alleles, one from each parent. Therefore, it is not impossible for a parent with the disease to have a child without it if the other parent possesses one functional allele of the gene for PAH. Yet, a child from two parents with PKU will inherit two mutated alleles every time, and therefore the disease.
Phenylketonuria can exist in mice, which have been extensively used in experiments into an effective treatment for it.[8] The macaque monkey's genome was recently sequenced, and the gene encoding phenylalanine hydroxylase was found to have the same sequence that, in humans, would be considered the PKU mutation.[9]
Tetrahydrobiopterin-deficient hyperphenylalaninemia
A rarer form of hyperphenylalaninemia occurs when PAH is normal, but there is a defect in the biosynthesis or recycling of the cofactor tetrahydrobiopterin (BH4) by the patient.[10] This cofactor is necessary for proper activity of the enzyme. The coenzyme (called biopterin) can be supplemented as treatment.
Levels of dopamine can be used to distinguish between these two types. Tetrahydrobiopterin is required to convert phenylalanine to tyrosine, but it is also required to convert tyrosine to L-DOPA (via the enzyme tyrosine hydroxylase), which in turn is converted to dopamine. Low levels of dopamine lead to high levels of prolactin. By contrast, in classical PKU, prolactin levels would be relatively normal. Tetrahydrobiopterin deficiency can be caused by defects in four different genes. These types are known as HPABH4A, HPABH4B, HPABH4C, and HPABH4D.[11]
Metabolic pathways
The enzyme phenylalanine hydroxylase normally converts the amino acid phenylalanine into the amino acid tyrosine. If this reaction does not take place, phenylalanine accumulates and tyrosine is deficient. Excessive phenylalanine can be metabolized into phenylketones through the minor route, a transaminase pathway with glutamate. Metabolites include phenylacetate, phenylpyruvate and phenethylamine.[12] Elevated levels of phenylalanine in the blood and detection of phenylketones in the urine is diagnostic, however most patients are diagnosed via newborn screening.
Phenylalanine is a large, neutral amino acid (LNAA). LNAAs compete for transport across the blood-brain barrier (BBB) via the large neutral amino acid transporter (LNAAT). If phenylalanine is in excess in the blood, it will saturate the transporter. Excessive levels of phenylalanine tend to decrease the levels of other LNAAs in the brain. However, as these amino acids are necessary for protein and neurotransmitter synthesis, Phe buildup hinders the development of the brain, causing mental retardation.[13]
Treatment
If PKU is diagnosed early enough, an affected newborn can grow up with normal brain development, but only by managing and controlling Phe levels through diet, or a combination of diet and medication. Optimal health ranges (or "target ranges") are between 120 and 360 µmol/L, and aimed to be achieved during at least the first 10 years.[14] When Phe cannot be metabolized by the body, abnormally high levels accumulate in the blood and are toxic to the brain. When left untreated, complications of PKU include severe mental retardation, brain function abnormalities, microcephaly, mood disorders, irregular motor functioning, and behavioral problems such as ADHD.
All PKU patients must adhere to a special diet low in Phe for optimal brain development. "Diet for life" has become the standard recommended by most experts. The diet requires severely restricting or eliminating foods high in Phe, such as meat, chicken, fish, eggs, nuts, cheese, legumes, milk and other dairy products. Starchy foods, such as potatoes, bread, pasta, and corn, must be monitored. Infants may still be breastfed to provide all of the benefits of breastmilk, but the quantity must also be monitored and supplementation for missing nutrients will be required. The sweetener aspartame, present in many diet foods and soft drinks, must also be avoided, as aspartame consists of two amino acids: phenylalanine and aspartic acid.
Supplementary infant formulas are used in these patients to provide the amino acids and other necessary nutrients that would otherwise be lacking in a low-phenylalanine diet. As the child grows up these can be replaced with pills, formulas, and specially formulated foods. (Since Phe is necessary for the synthesis of many proteins, it is required for appropriate growth, but levels must be strictly controlled in PKU patients.) In addition, tyrosine, which is normally derived from phenylalanine, must be supplemented.
The oral administration of tetrahydrobiopterin (or BH4) (a cofactor for the oxidation of phenylalanine) can reduce blood levels of this amino acid in certain patients.[15][16] The company BioMarin Pharmaceutical has produced a tablet preparation of the compound sapropterin dihydrochloride (Kuvan), which is a form of tetrahydrobiopterin. Kuvan is the first drug that can help BH4-responsive PKU patients (defined among clinicians as about 1/2 of the PKU population) lower Phe levels to recommended ranges.[17] Working closely with a dietitian, some PKU patients who respond to Kuvan may also be able to increase the amount of natural protein they can eat.[18] After extensive clinical trials, Kuvan has been approved by the FDA for use in PKU therapy. Some researchers and clinicians working with PKU are finding Kuvan a safe and effective addition to dietary treatment and beneficial to patients with PKU.[19][20]
Several other therapies are currently under investigation, including gene therapy, large neutral amino acids, and enzyme substitution therapy with phenylalanine ammonia lyase (PAL). In the past, PKU-affected people were allowed to go off diet after approximately eight, then 18 years of age. Today, most physicians recommend PKU patients must manage their Phe levels throughout life.
Maternal phenylketonuria

For women with PKU, it is essential for the health of their children to maintain low Phe levels before and during pregnancy.[21] Though the developing fetus may only be a carrier of the PKU gene, the intrauterine environment can have very high levels of phenylalanine, which can cross the placenta. The child may develop congenital heart disease, growth retardation, microcephaly and mental retardation as a result.[22] PKU-affected women themselves are not at risk of additional complications during pregnancy.
In most countries, women with PKU who wish to have children are advised to lower their blood Phe levels (typically to between 2 and 6 micromol/deciliter) before they become pregnant, and carefully control their levels throughout the pregnancy. This is achieved by performing regular blood tests and adhering very strictly to a diet, in general monitored on a day-to-day basis by a specialist metabolic dietitian. In many cases, as the fetus' liver begins to develop and produce PAH normally, the mother's blood Phe levels will drop, requiring an increased intake to remain within the safe range of 2-6 micromol/dL. The mother's daily Phe intake may double or even triple by the end of the pregnancy, as a result. When maternal blood Phe levels fall below 2 micromol/dL, anecdotal reports indicate that the mothers may suffer adverse effects, including headaches, nausea, hair loss, and general malaise. When low phenylalanine levels are maintained for the duration of pregnancy, there are no elevated levels of risk of birth defects compared with a baby born to a non-PKU mother.[23] Babies with PKU may drink breast milk, while also taking their special metabolic formula. Some research has indicated an exclusive diet of breast milk for PKU babies may alter the effects of the deficiency, though during breastfeeding the mother must maintain a strict diet to keep her Phe levels low. More research is needed. US scientist announced in June 2010 that they would be conducting a thorough investigation on the mutation of genes in the human genome. Their top priority is PKU, as it has become increasingly common, and sufferers often bear children who will be carriers of the recessive gene, and may themselves live past the age of sixty.
Incidence
The mean incidence of PKU varies widely in different human populations. In Turkey, 1 in 2600 births (the highest rate in the world) show PKU; in Ireland, 1 in 4,500,[24] 1 in 13,000 in Norway,[25] and fewer than one in 100,000 in Finland.[26] In the United States, about 1 in 15,000 births show classical PKU.[27] The incidence is relatively high in Italy, China, and Yemen.[27]
See also
- Hyperphenylalanemia
- Lofenalac
- Tetrahydrobiopterin deficiency
References
- ^ James, William D.; Berger, Timothy G. (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. ISBN 0-7216-2921-0.
- ^ Gonzalez, Jason; Willis, Monte S. (Feb. 2010). "Ivar Asbjorn Folling Discovered Phenylketonuria (PKU)". Lab medicine 41 (2): 118–119.
- ^ Folling, A. (1934). "Ueber Ausscheidung von Phenylbrenztraubensaeure in den Harn als Stoffwechselanomalie in Verbindung mit Imbezillitaet". Ztschr. Physiol. Chem. 227: 169–176.
- ^ Centerwall, S. A. & Centerwall, W. R. (2000). "The discovery of phenylketonuria: the story of a young couple, two affected children, and a scientist". Pediatrics 105 (1 Pt 1): 89–103. doi:10.1542/peds.105.1.89. PMID 10617710. http://pediatrics.aappublications.org/cgi/content/full/105/1/89.
- ^ Mayo Clinic Staff (2007-12-20). "Phenylketonuria (PKU)". Mayo Clinic. http://www.mayoclinic.com/health/phenylketonuria/DS00514/DSECTION=1. Retrieved 2008-03-13.
- ^ Sarafoglou, Kyriakie; Hoffmann, Georg F.; Roth, Karl S., eds. Pediatric Endocrinology and Inborn Errors of Metabolism. New York: McGraw Hill Medical. p. 26.
- ^ http://www.genenames.org Phenylalanine hydroxylase (PAH) gene summary, retrieved September 8, 2006
- ^ Oh, H. J., Park, E. S., Kang, S., Jo, I., Jung, S. C. (2004). "Long-Term Enzymatic and Phenotypic Correction in the Phenylketonuria Mouse Model by Adeno-Associated Virus Vector-Mediated Gene Transfer". Pediatric Research 56 (2): 278–284. doi:10.1203/01.PDR.0000132837.29067.0E. PMID 15181195. http://www.pedresearch.org/cgi/content/full/56/2/278.
- ^ Gibbs, Richard A., Jeffrey Rogers, Michael G. Katze, Roger Bumgarner, George M. Weinstock, Elaine R. Mardis et al. (April 2007). "Evolutionary and Biomedical Insights from the Rhesus Macaque Genome". Science 316 (5822): 222–234. doi:10.1126/science.1139247. PMID 17431167. http://www.sciencemag.org/cgi/content/full/316/5822/222. Retrieved 2008-02-26.
- ^ Surtees, R., Blau, N. (2000). "The neurochemistry of phenylketonuria". European Journal of Pediatrics 169: S109–S113. doi:10.1007/PL00014370. PMID 11043156.
- ^ Online 'Mendelian Inheritance in Man' (OMIM) 261640
- ^ Michals, K., Matalon, R. (1985). "Phenylalanine metabolites, attention span and hyperactivity". American Journal of Clinical Nutrition 42 (2): 361–5. PMID 4025205.
- ^ Pietz, J., Kreis, R., Rupp, A., Mayatepek, E., Rating, D., Boesch, C., Bremer, H. J. (1999). "Large neutral amino acids block phenylalanine transport into brain tissue in patients with phenylketonuria". Journal of Clinical Investigation 103 (8): 1169–1178. doi:10.1172/JCI5017. PMC 408272. PMID 10207169. http://www.jci.org/cgi/content/full/103/8/1169.
- ^ Chapter 55, page 255 in:Behrman, Richard E.; Kliegman, Robert; Nelson, Waldo E.; Karen Marcdante; Jenson, Hal B. (2006). Nelson essentials of pediatrics. Elsevier/Saunders. ISBN 1-4160-0159-X.
- ^ Burton, BK; Kar S, Kirkpatrick P (2008). "Fresh from the Pipeline: Sapropterin". Nature Reviews Drug Discovery 7 (3): 199–200. doi:10.1038/nrd2540. http://www.nature.com/nrd/journal/v7/n3/full/nrd2540.html.
- ^ Michals-Matalon K (2008). "Sapropterin dihydrochloride, 6-R-L-erythro-5,6,7,8-tetrahydrobiopterin, in the treatment of phenylketonuria". Expert Opin Investig Drugs 17 (2): 245–251. doi:10.1517/13543784.17.2.245. PMID 18230057.
- ^ Burton BK, Grange DK, Milanowski A, Vockley G, Feillet F, Crombez EA et al. (2007). "The response of patients with phenylketonuria and elevated serum phenylalanine to treatment with oral sapropterin dihydrochloride (6R-tetrahydrobiopterin): a phase II, multicentre, open-label, screening study". Journal of Inherited Metabolic Disorders 30 (5): 700–707. doi:10.1007/s10545-007-0605-z. PMID 17846916. http://www.nature.com/nrd/journal/v7/n3/full/nrd2540.html.
- ^ Levy H, Burton B, Cederbaum S et al. (2007). "Recommendations for evaluation of responsiveness to tetrahydrobiopterin (BH(4)) in phenylketonuria and its use in treatment". Mol Genet Metab 92 (4): 287–291. doi:10.1016/j.ymgme.2007.09.017. PMID 18036498.
- ^ Levy HL, Milanowski A, Chakrapani A, Cleary M, Lee P, Trefz FK et al. (2007). "Efficacy of sapropterin dihydrochloride (tetrahydrobiopterin, 6R-BH4) for reduction of phenylalanine concentration in patients with phenylketonuria: a phase III randomised placebo-controlled study". Lancet 370 (9586): 504–510. doi:10.1016/S0140-6736(07)61234-3. PMID 17693179.
- ^ Lee P, Treacy E, Crombez E et al. (2008). "Safety and efficacy of 22 weeks of treatment with sapropterin dihydrochloride in patients with phenylketonuria". Am J Med Genet 146A (22): 2851–2859. doi:10.1002/ajmg.a.32562. PMID 18932221.
- ^ Lee, P.J., Ridout, D., Walker, J.H., Cockburn, F., (2005). "Maternal phenylketonuria: report from the United Kingdom Registry 1978–97". Archives of Disease in Childhood 90 (2): 143–146. doi:10.1136/adc.2003.037762. PMC 1720245. PMID 15665165. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1720245..
- ^ Rouse, B., Azen, B., Koch, R., Matalon, R., Hanley, W., de la Cruz, F., Trefz, F., Friedman, E., Shifrin, H. (1997). "Maternal phenylketonuria collaborative study (MPKUCS) offspring: Facial anomalies, malformations, and early neurological sequelae". American Journal of Medical Genetics 69 (1): 89–95. doi:10.1002/(SICI)1096-8628(19970303)69:1<89::AID-AJMG17>3.0.CO;2-K. PMID 9066890.
- ^ lsuhsc.edu Genetics and Louisiana Families
- ^ DiLella, A. G., Kwok, S. C. M., Ledley, F. D., Marvit, J., Woo, S. L. C. (1986). "Molecular structure and polymorphic map of the human phenylalanine hydroxylase gene". Biochemistry 25 (4): 743–749. doi:10.1021/bi00352a001. PMID 3008810.
- ^ www.rikshospitalet.no
- ^ Guldberg, P., Henriksen, K. F., Sipila, I., Guttler, F., de la Chapelle, A. (1995). "Phenylketonuria in a low incidence population: molecular characterization of mutations in Finland". J. Med. Genet 32 (12): 976–978. doi:10.1136/jmg.32.12.976. PMC 1051781. PMID 8825928. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1051781.
- ^ a b http://emedicine.medscape.com/article/947781-overview
External links
- Phenylketonuria at the Open Directory Project
- Canadian PKU and Allied Disorders
- GeneReviews/NCBI/NIH/UW entry on Phenylalanine Hydroxylase Deficiency
- National Society for Phenylketonuria
- National PKU News
- Videos, Blogs, PKU Community
- Online PKU Community
Inborn error of amino acid metabolism (E70–E72, 270) K→acetyl-CoA HypertryptophanemiaG Glutamate/glutamineG→fumaratePhenylketonuriaType II tyrosinemia · Type III tyrosinemia/Hawkinsinuria · Alkaptonuria/Ochronosis · Type I tyrosinemiaTransport/
IE of RTTOther Categories:- Mental retardation
- Autosomal recessive disorders
- Amino acid metabolism disorders
- Skin conditions resulting from errors in metabolism

Wikimedia Foundation. 2010.