- 3-hydroxy-3-methylglutaryl-CoA lyase deficiency
Infobox_Disease
Name = PAGENAME
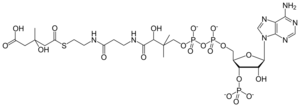
Caption =HMG-CoA
DiseasesDB = 29826
ICD10 =
ICD9 =
ICDO =
OMIM = 246450
MedlinePlus =
eMedicineSubj =
eMedicineTopic =
MeshID = C0080473-hydroxy-3-methylglutaryl-CoA lyase deficiency also referred to as HMG-CoA lyase deficiency or Hydroxymethylglutaric aciduria, is an uncommon inherited disorder in which the body cannot properly process the
amino acid leucine. Additionally, the disorder prevents the body from makingketone s, which are used for energy during fasting.Presentation
This disorder usually appears within the first year of life. The signs and symptoms of
HMG-CoA lyase deficiency includevomiting ,dehydration ,lethargy ,convulsion s, andcoma . When episodes occur in an infant or child,blood sugar becomes extremely low (hypoglycemia ), and harmful compounds can build up and cause the blood to become too acidic (metabolic acidosis ). These episodes are often triggered by an infection, fasting, strenuous exercise, or sometimes other types of stress.Differential diagnosis
This condition is sometimes mistaken for
Reye syndrome , a severe disorder that develops in children while they appear to be recovering from viral infections such aschicken pox orflu . Most cases of Reye syndrome are associated with the use of aspirin during these viral infections.Cause
Mutation s in the "HMGCL"gene cause 3-hydroxy-3-methylglutaryl-CoA lyase deficiency. Theenzyme made by the "HMGCL" gene plays an essential role in breaking down dietary proteins and fats for energy. Specifically, the enzyme is responsible for processing leucine, an amino acid that is part of many proteins. This enzyme also produces ketones during the breakdown of fats. If a mutation in the "HMGCL" gene reduces or eliminates the activity of this enzyme, the body is unable to process leucine or make ketones properly. A lack of ketones leads to hypoglycemia, and compounds calledorganic acids (which are formed as products of amino acid and fat breakdown) can cause the blood to become too acidic. Metabolic acidosis and hypoglycemia impair tissue function, especially in thecentral nervous system .Incidence
This is a rare condition that has been reported in fewer than 100 individuals throughout the world. This condition is inherited in an
autosomal recessive pattern.ee also
*
3-hydroxy-3-methylglutaryl-CoA lyase References
"This article incorporates public domain text from [http://ghr.nlm.nih.gov The U.S. National Library of Medicine] "
External links
* [http://www.oaanews.org Organic Acidemia Association]
*
*http://ghr.nlm.nih.gov/condition=3hydroxy3methylglutarylcoenzymealyasedeficiency;jsessionid=3329E3ED085B1A6E5BE651FC9159D676
*http://www.newbornscreening.info/Parents/organicaciddisorders/HMGCoA.html

Wikimedia Foundation. 2010.