- 3-Methylglutaconic aciduria
-
3-Methylglutaconic aciduria Classification and external resources 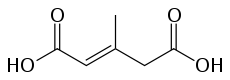
3-methylglutaconic acidDiseasesDB 29831 29297 29832 29833 3-Methylglutaconic aciduria (MGA) is used to describe at least five different disorders that impair the body's ability to make energy in the mitochondria. As a result of this impairment, 3-methylglutaconic acid and 3-methylglutaric acid build up and can be detected in the urine.
3-Methylglutaconic acid is classified as an organic acid. The double carboxylic acid functions are the principal cause of the strength of this acid. 3-methylglutaconic acid can be detected by the presence of the acid function and the double connection that involves reactivity with some specific substances.
Contents
Classification
There are currently 5 known subgroups of MGA; MGA type I,II,III,IV & V.
Type OMIM Gene Locus Also known as/Description Genetics Type I 250950 AUH Chr.9 3-Methylglutaconic aciduria type I, 3-Methylglutaconic acidemia or 3-Methylglutaconyl-CoA Hydratase Deficiency[1] Mutations in the AUH gene cause 3-methylglutaconic aciduria type I. This gene provides instructions for producing 3-methylglutaconyl-CoA hydratase, an enzyme that is involved in processing the amino acid leucine. This amino acid is broken down in the mitochondria during the process of energy production. A deficiency of this enzyme leads to a buildup of 3-methylglutaconic acid, which is eliminated in the urine. Researchers believe that other genes or environmental factors also contribute to the development of this disorder. Type II 302060 TAZ Xq28 Barth syndrome (BTHS), 3-Methylglutaconic aciduria type II or Cardiomyopathy-neutropenia syndrome Mutations in the TAZ gene cause 3-methylglutaconic aciduria type II (Barth syndrome). This gene provides instructions for making a protein called tafazzin. This protein plays a critical role in maintaining the levels of a particular lipid, called cardiolipin, located in the inner membrane of the mitochondria. A lack of tafazzin results in abnormalities in the structure and function of mitochondria, leading to the heart defects and other problems seen in this disorder. Type III 258501 OPA3 19q13.2-q13.3 3-Methylglutaconic aciduria type III or Costeff syndrome[2] Mutations in the OPA3 gene cause 3-methylglutaconic aciduria type III. This gene carries instructions for making a protein that is also found in mitochondria, but whose function is unknown. Researchers have suggested that cells with a defective OPA3 protein are more susceptible to a process that eliminates damaged or unneeded cells (programmed cell death called apoptosis). Type IV 250951 ? ? 3-Methylglutaconic aciduria type IV Type V 610198 DNAJC19 3q26.3 3-Methylglutaconic aciduria type V The characteristic features of 3-methylglutaconic aciduria type I include speech delay, delayed development of both mental and motor skills (psychomotor delay), elevated levels of acid in the blood and tissues (metabolic acidosis), abnormal muscle tone (dystonia), and spasms and weakness affecting the arms and legs (spastic quadriparesis). Fewer than 20 cases of 3-methylglutaconic aciduria type I have been reported.
Barth syndrome is a common name for 3-methylglutaconic aciduria type II. The main features of Barth syndrome include a weakened and enlarged heart (dilated cardiomyopathy), recurrent infections due to low numbers of white blood cells (neutropenia), skeletal problems, and delayed growth. The incidence of 3-methylglutaconic aciduria type II is approximately 1 in 200,000 male infants.
Costeff optic atrophy syndrome is another name for 3-methylglutaconic aciduria type III. This disorder is characterized mainly by the degeneration of the optic nerves, which carry information from the eyes to the brain. Sometimes other nervous system problems occur, such as an inability to maintain posture, poor muscle tone, the development of certain involuntary movements (extrapyramidal dysfunction), and a general decrease in brain function (cognitive deficit). The incidence of 3-methylglutaconic aciduria type III is about 1 in 10,000 newborns in the Iraqi Jewish population. This disorder is extremely rare in all other populations.
The signs and symptoms of 3-methylglutaconic aciduria type IV are variable and overlap with types I-III. The incidence of 3-methylglutaconic aciduria type IV is unknown.
Epidemiology
3-Methylglutaconic aciduria, seems to be most prevalent amongst the Jewish population of Iraq. However, a high concentration of one type is found in the Saguenay-Lac-Saint-Jean region of Canada. This tends to show that the disease is more frequent in insular areas where there is more chance that both parents be carriers, a higher birth rate, and higher number of congenital marriages. As all types of 3-Methylglutaconic aciduria are known to be genetic diseases and show a recessive pattern it is likely that congenital marriages where both partners are carriers increase the chance to have a baby with the condition.
Inheritance patterns
The inheritance patterns of 3-methylglutaconic aciduria differ depending on the gene involved.
- Types I and III are inherited in an autosomal recessive pattern, which means two copies of the gene in each cell are altered. Most often, the parents of an individual with an autosomal recessive disorder are carriers of one copy of the altered gene but do not show signs and symptoms of the disorder.
- Type II is inherited in an X-linked recessive pattern. A condition is considered X-linked if the mutated gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a mutation must be present in both copies of the gene to cause the disorder. Males are affected by X-linked recessive disorders much more frequently than females. A striking characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
- The inheritance pattern of 3-methylglutaconic aciduria type IV is unknown.
References
External links
- GeneReviews/NCBI/NIH/UW entry on 3-Methylglutaconic Aciduria Type 3
- OMIM entries on 3-Methylglutaconic Aciduria Type 3
- 3-Methylglutaconic aciduria at NLM Genetics Home Reference
Inborn error of amino acid metabolism (E70–E72, 270) K→acetyl-CoA Lysine/straight chainMaple syrup urine disease · Isovaleric acidemia · 3-Methylcrotonyl-CoA carboxylase deficiency · 3-hydroxy-3-methylglutaryl-CoA lyase deficiency · 3-Methylglutaconic aciduria 1HypertryptophanemiaG G→pyruvate→citrateG→glutamate→
α-ketoglutarateGlutamate/glutamineG→fumarateType II tyrosinemia · Type III tyrosinemia/Hawkinsinuria · Alkaptonuria/Ochronosis · Type I tyrosinemiaTransport/
IE of RTTOther Tafazzin Other Paroxysmal nocturnal hemoglobinuria · Hyperphosphatasia with mental retardation syndromeTranslation Ribosome: Diamond–Blackfan anemia · FMR1 (Fragile X syndrome, Fragile X-associated tremor/ataxia syndrome, Premature ovarian failure 1)
Initiation factor: Leukoencephalopathy with vanishing white matter
snRNP: Retinitis pigmentosa 33Posttranslational modification Alzheimer's disease · Huntington's disease · Creutzfeldt–Jakob disease
chaperonins: 3-Methylglutaconic aciduria 5E1: X-linked spinal muscular atrophy 2
E3: Johanson–Blizzard syndrome · Von Hippel–Lindau disease · 3-M syndrome · Angelman syndrome
Deubiquitinating enzyme: Machado–Joseph disease · Aneurysmal bone cyst · Multiple familial trichoepithelioma 1Othersee also genetic translation, posttranslational modification
B structural (perx, skel, cili, mito, nucl, sclr) · DNA/RNA/protein synthesis (drep, trfc, tscr, tltn) · membrane (icha, slcr, atpa, abct, othr) · transduction (iter, csrc, itra), trfkCategories:- Phospholipid metabolism disorders

Wikimedia Foundation. 2010.