- Ehlers–Danlos syndrome
-
Ehlers–Danlos syndrome Classification and external resources 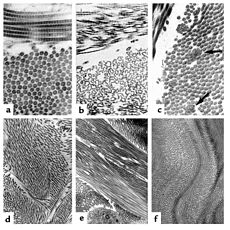
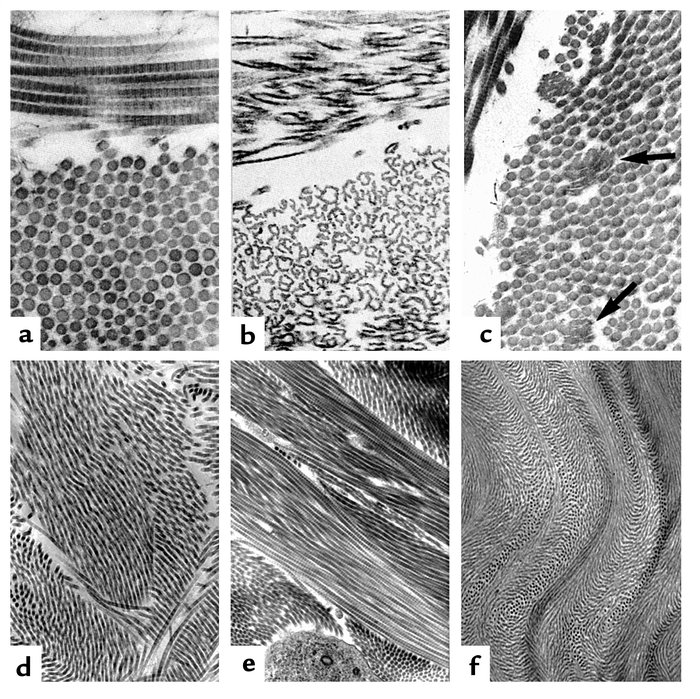
The collagen fibril and EDS. (a) Normal collagen fibrils are of uniform size and spacing. Fibrils from a patient with dermatosparaxis (b) show dramatic alterations in fibril morphology with severe effects on tensile strength of connective tissues. Patients with classical EDS (c) show composite fibrils. Fibrils from a TNX-deficient patient (d) are uniform in size and no composite fibrils are seen. TNX-null (e) fibrils are less densely packed and not as well aligned to neighboring fibrils.ICD-10 Q79.6 (ILDS Q82.817) ICD-9 756.83 MedlinePlus 001468 eMedicine derm/696 ped/654 MeSH D004535 Ehlers–Danlos syndrome (EDS) (also known as "Cutis hyperelastica"[1]) is a group of inherited connective tissue disorders, caused by a defect in the synthesis of collagen (Type I or III). The collagen in connective tissue helps tissues to resist deformation. In the skin, muscles, ligaments, blood vessels and visceral organs, collagen plays a very significant role and with increased elasticity, secondary to abnormal collagen, pathology results. Depending on the individual mutation, the severity of the syndrome can vary from mild to life-threatening. There is no cure, and treatment is supportive, including close monitoring of the digestive, excretory and particularly the cardiovascular systems. Corrective surgery may help with the frequent injuries that may develop in certain types of EDS, although the condition means that extra caution is advised and special practices observed.[2]
The syndrome is named after two doctors, Edvard Ehlers of Denmark, and Henri-Alexandre Danlos of France, who identified it at the turn of the 20th century.[3]
Contents
Classification
In the past, there were more than 10 recognized types of Ehlers–Danlos syndrome. In 1997, researchers proposed a simpler classification that reduced the number of major types to six and gave them descriptive names.[4] These six major types are listed here. Other types of the condition may exist, but they have been reported only in single families or are not well characterized. Except for hypermobility, the specific mutations involved have been identified and they can be precisely identified by genetic testing; this is valuable due to a great deal of variation in individual presentation of symptoms which may confuse classification of individuals on purely symptomatic basis. In order of prevalence in the population, they are:
Name Number Description OMIM Gene(s) Hypermobility type 3 Affects 1 in 10,000 to 15,000 and is caused by an autosomal dominant or autosomal recessive mechanism. Mutations in either of two separate genes (which are also involved in Vascular EDS and Tenascin-X deficiency EDS, respectively) may lead to this variant. Extreme hypermobility is the hallmark of this type. 130020 COL3A1, TNXB Classical types 1 & 2 Affects approximately 1 in 20,000 to 50,000 people. It is caused by autosomal dominant mechanism and affects type-V collagen, as well as type I. Type 1 typically presents with severe skin involvement, and type 2 presents with mild to moderate skin involvement. 130000, 130010 COL5A1, COL5A2, COL1A1 Vascular type 4 Is an autosomal dominant defect in the type-III collagen synthesis; affecting approximately 1 in 100,000 to 250,000 people. The vascular type is considered one of the more serious forms of Ehlers–Danlos syndrome because blood vessels and organs are more prone to tearing (rupture). Many patients with EDS type 4 express a characteristic facial appearance (large eyes, small chin, thin nose and lips, lobeless ears), have a small stature with a slim build, and typically have thin, pale, translucent skin (veins can usually be seen on the chest and abdomen). About one in four people with vascular type EDS develop a significant health problem by age 20 and more than 80 percent develop life-threatening complications by age 40. 130050 COL3A1 Kyphoscoliosis type 6 Is an autosomal recessive defect due to deficiency of an enzyme called lysyl hydroxylase; it is very rare, with fewer than 60 cases reported. The kyphoscoliosis type is characterised by progressive curvature of the spine (scoliosis), fragile eyes, and severe muscle weakness. 225400, 229200 PLOD1 Arthrochalasis types 7A & B Is also very rare, with about 30 cases reported. It affects type-I collagen. The arthrochalasia type is characterised by very loose joints and dislocations involving both hips. 130060 COL1A1, COL1A2 Dermatosparaxis type 7C Also very rare, with about 10 cases reported. The dermatosparaxis type is characterised by extremely fragile and sagging skin. 225410 ADAMTS2 Other types
Although the above classifications are well defined, it is rare for a case to fit neatly in a single category, and cross-over symptoms lead to under-diagnosis or mis-diagnosis. So patients should not rely on the "fact" of having a certain type of EDS if cross-over symptoms are evident and might be life-threatening.
"The large number of distinct types of the Ehlers–Danlos syndrome that have already been identified indicates great heterogeneity, but clearly that heterogeneity is not exhausted by the present classification." [1] Forms of EDS within this category may present with soft, mildly stretchable skin, shortened bones, chronic diarrhea, joint hypermobility and dislocation, bladder rupture, or poor wound healing. Inheritance patterns within this group include X-linked recessive, autosomal dominant, and autosomal recessive. Examples of types of related syndromes other than those above reported in the medical literature include:
- 305200 – Type 5
- 130080 – Type 8 - unspecified gene, locus 12p13
- 225310 – Type 10 - unspecified gene, locus 2q34
- 608763 – Beasley-Cohen type
- 130070 – Progeroid form - B4GALT7
- 606408 – Due to Tenascin-X deficiency - TNXB
- 130090 – Type unspecified
Signs and symptoms

 Individual with EDS displaying hypermobile joints.
Individual with EDS displaying hypermobile joints.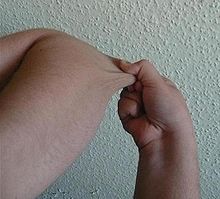 Individual with EDS displaying skin hyperelasticity
Individual with EDS displaying skin hyperelasticity
Signs vary widely based on which type of EDS the patient has. In each case, however, the signs are ultimately due to faulty or reduced amounts of collagen. EDS most typically affects the joints, skin, and blood vessels, the major signs and symptoms include:
- Loose, unstable joints that are prone to: sprain, dislocation, subluxation (partial dislocation) and hyperextension (double jointedness) [5]
- Early onset of osteoarthritis
- Easy bruising
- Dysautonomia typically accompanied by Valvular heart disease (such as mitral valve prolapse, which creates an increased risk for infective endocarditis during surgery, as well as possibly progressing to a life-threatening degree of severity of the prognosis of mitral valve prolapse) [6]
- Chronic fatigue
- Flat feet
- High and narrow palate, resulting in dental crowding
- Vulnerability to chest and sinus infections
- Fragile blood vessels resulting from cystic medial necrosis with tendency towards aneurysm (even abdominal aortic aneurysm)
- Velvety-smooth skin which may be stretchy, is often translucent, and can contribute to underestimations of chronological age
- Abnormal wound healing and scar formation
- Low muscle tone and muscle weakness
- Migraines and headaches, including postural headaches from spontaneous intracranial hypotension
- Myalgia and arthralgia[7]
Other, less common signs and complications may include:
- Osteopenia (low bone density)
- Talipes equinovarus (club foot), especially in the Vascular type
- Deformities of the spine, such as: Scoliosis (curvature of the spine), Kyphosis (a thoracic hump), Tethered spinal cord syndrome, Occipitoatlantoaxial hypermobility[8]
- Arnold-Chiari malformation (brain disorder)[9]
- Functional bowel disorders (functional gastritis, irritable bowel syndrome)
- Gastroparesis
- Dental issues, including early-onset periodontitis
- Nerve compression disorders (carpal tunnel syndrome, acroparesthesia, neuropathy) [10]
- Vascular skin conditions: Raynaud's phenomenon, Livedo reticularis
- Blue sclera
- Swan neck deformity of the fingers [11]
- Insensitivity to local anesthetics.[12]
- Premature rupture of membranes during pregnancy [13]
- Platelet aggregation failure (platelets do not clump together properly) [14]
- Weak muscle tone (hypotonia) in infancy, which can delay the development of motor skills such as sitting, standing, and walking
- Arterial/intestinal/uterine fragility or rupture
Because it is often undiagnosed or misdiagnosed in childhood, some instances of Ehlers–Danlos syndrome have been mischaracterized as child abuse.[15] The pain associated with this condition is a serious complication.
Genetics
Mutations in the following can cause Ehlers–Danlos syndrome:
Mutations in these genes usually alter the structure, production, or processing of collagen or proteins that interact with collagen. Collagen provides structure and strength to connective tissue throughout the body. A defect in collagen can weaken connective tissue in the skin, bones, blood vessels, and organs, resulting in the features of the disorder.
Inheritance patterns depend on the type of Ehlers–Danlos syndrome. Most forms of the condition are inherited in an autosomal dominant pattern, which means only one of the two copies of the gene in question must be altered to cause the disorder. The minority are inherited in an autosomal recessive pattern, which means both copies of the gene must be altered for a person to be affected by the condition. It can also be an individual (de novo or "sporadic") mutation. Please refer to the summary for each type of Ehlers–Danlos syndrome for a discussion of its inheritance pattern.
Diagnosis
A diagnosis can be made by clinical observation. Both DNA and biochemical studies can be used to help identify affected individuals. In some cases, a skin biopsy has been found to be useful in confirming a diagnosis. Unfortunately, these tests are not sensitive enough to identify all individuals with EDS. If there are multiple affected individuals in a family, it may be possible to perform prenatal diagnosis using a DNA information technique known as a linkage study.
Differential diagnosis
There are several disorders that have some of the characteristics of Ehlers–Danlos syndrome. For example, in cutis laxa the skin is loose, hanging, and wrinkled. In EDS, the skin can be pulled away from the body but is elastic and returns to normal when let go. In Marfan syndrome, the joints are very mobile and similar cardiovascular complications occur. In the past, Menkes disease, a copper metabolism disorder, was thought to be a form of Ehlers–Danlos syndrome. Because of these similar disorders, a correct diagnosis is very important.[16]
Management
There is no cure for Ehlers Danlos Syndrome. The treatment is supportive. Close monitoring of the cardiovascular system, physical therapy, occupational therapy, and orthopedic instruments (e.g., wheelchairs, bracing) may be helpful. One should avoid activities that cause the joint to lock or overextend.
A physician may prescribe bracing to stabilize joints. Surgical repair of joints may be necessary at some time. Physicians may also consult a physical and/or occupational therapist to help strengthen muscles and to teach people how to properly use and preserve their joints. To decrease bruising and improve wound healing, some patients have responded to ascorbic acid (vitamin C). [17]
In general, medical intervention is limited to symptomatic therapy. Prior to pregnancy, patients with EDS should have genetic counseling. Children with EDS should be provided with information about the disorder, so they can understand why contact sports and certain other physically stressful activities should be avoided. Children should be taught early on that demonstrating the unusual positions they can maintain due to loose joints should not be done as this may cause early degeneration of the joints. Family members, teachers and friends should be provided with information about EDS so they can accept and assist the child as necessary.
Prognosis
The outlook for individuals with EDS depends on the type of EDS with which they have been diagnosed. Symptoms vary in severity, even within one sub-type, and the frequency of complications changes on an individual basis. Some individuals have negligible symptoms while others are severely restricted in their daily life. Extreme joint instability, pain, and spinal deformities may limit a person's mobility. Most individuals will have a normal lifespan. However, those with blood vessel involvement have an increased risk of fatal complications.
EDS is a lifelong condition. Affected individuals may face social obstacles related to their disease on a daily basis. Some people with EDS have reported living with fears of significant and painful ruptures, their condition worsening, becoming unemployed due to physical and emotional burdens, and social stigmatization in general.
Epidemiology
Ehlers–Danlos syndrome is an inherited disorder estimated to occur in about 1 in 5000 births worldwide. Initially, prevalence estimates ranged from 1 in 250,000 to 1 in 500,000 people, but these estimates were soon found to be vastly inaccurate as the disorder received further study and medical professionals became more adept at accurately diagnosing EDS. In fact, many experts now believe that Ehlers–Danlos syndrome may be far more common than the currently accepted estimate due to the wide range of severities in which the disorder presents.[18] However, the prevalence of the six types differs dramatically. The most commonly occurring type is the hypermobility type, followed by the classical type. The other types of Ehlers–Danlos syndrome are very rare. For example, fewer than 10 infants and children with the dermatosparaxis type have been described worldwide. Ehlers–Danlos affects both males and females of all racial and ethnic backgrounds, although some types are more common among certain groups than others.
Society and culture
- Several celebrities have EDS:
- A man with EDS who has the stretchiest skin in the world appears in an episode entitled Rubber Band Man in the show "Stan Lee's Superhumans".
- Actress Cherylee Houston has type 3 EDS and uses a wheelchair; she made history by becoming Coronation Street's first full-time disabled actress.[19]
- The condition may have contributed to the virtuoso violinist Niccolò Paganini's skill as he was able to play wider fingerings than the normal violinist.[20]
- Eric the Midget, of the Howard Stern show has a rare form of Ehlers-Danlos syndrome.
- Pianist Evan Petrone's video "Backwards Piano" went viral and gained much popularity after it was released that he suffers from Ehlers Danlos Syndrome Type 3. [21]
- Musician Noah Baerman has been a long-time supporter of EDS research, selling his first CD, Patch Kit, to benefit the Ehlers–Danlos National Foundation.
- The condition is mentioned in the song "Dorsal Horn Concerto" by the British comedy band the Amateur Transplants.[22]
- The condition has been mentioned in many television shows, including:
- CSI New York in the first season episode, "Blood, Sweat and Tears."
- FOX series Bones in the season 4 episode "The Perfect Pieces in the Purple Pond."
- House in the episodes "House's Head" (Season 4, Episode 15) and "The Dig (House)" (Season 7, Episode 18).
- The Law & Order episode "Dignity" (Season 20, Episode 5). In the episode, a third-trimester fetus is diagnosed with the condition and is slated for abortion; shortly later, the doctor who is due to perform the abortion is murdered in church.
- The subject of the Season 7 premiere of the A&E series Intervention, Linda, claims to have EDS.
In other animals
Ehlers–Danlos-like syndromes have been shown to be hereditary in Himalayan cats, some domestic shorthair cats, and in certain breeds of cattle. It is seen as a sporadic condition in domestic dogs.
DSLD: Degenerative Suspensory Ligament Desmitis is a similar condition now being researched in all breeds of horses. Though it was originally notated in the Peruvian Paso and thought to be a condition of overwork and older age, the disease is being recognized in all age groups and all activity levels. It has even been noted in newborn foals. The latest research has led to the renaming of the disease after the possible systemic and hereditary components now being delineated by the University of Georgia. Equine Systemic Proteoglycan Accumulation.[23][24]
See also
- Abdominal aortic aneurism
- Arnold-Chiari malformation
- Joint dislocation
- Dural ectasia
- Dysautonomia
- Gorlin sign
- Hypermobility
- Marfan syndrome
- mitral valve prolapse
- Postural orthostatic tachycardia syndrome
- Subluxations
- Tethered spinal cord syndrome
- Tricuspid regurgitation
- Wrinkly skin syndrome
References
- ^ James, William D.; Berger, Timothy G.; Elston, Dirk M. (2006). Andrews' diseases of the skin: clinical dermatology (10th ed.). Philadelphia: Saunders. p. 512. ISBN 978-0-7216-2921-6.
- ^ http://arthritisinsight.com/medical/surgery/eds.html
- ^ "Uncovered: How U.S. Health System Can Fail Even the Insured --- A Woman Endures 16-Month Odyssey To Get a Diagnosis", John Carreyrou, Wall Street Journal, November 16, 2007
- ^ Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ (1998). "Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK)". Am J Med Genet 77 (1): 31–7. doi:10.1002/(SICI)1096-8628(19980428)77:1<31::AID-AJMG8>3.0.CO;2-O. PMID 9557891.
- ^ Lawrence EJ (2005). "The clinical presentation of Ehlers-Danlos syndrome". Adv Neonatal Care 5 (6): 301–14. doi:10.1016/j.adnc.2005.09.006. PMID 16338669.
- ^ http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=eds3
- ^ Gedalia, A; Press, J; Klein, M; Buskila, D (1993). "Joint hypermobility and fibromyalgia in schoolchildren.". Annals of the Rheumatic Diseases 52 (7): 494. doi:10.1136/ard.52.7.494. PMC 1005086. PMID 8346976. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1005086.
- ^ Milhorat TH, Bolognese PA, Nishikawa M, McDonnell NB, Francomano CA (December 2007). "Syndrome of occipitoatlantoaxial hypermobility, cranial settling, and chiari malformation type I in patients with hereditary disorders of connective tissue". Journal of Neurosurgery. Spine 7 (6): 601–9. doi:10.3171/SPI-07/12/601. PMID 18074684.
- ^ http://www.northshorelij.com/workfiles/chiari/J%20Neurosurg%20Spine%20article%20Dec%2007.pdf
- ^ http://www.answers.com/topic/ehlers-danlos-syndrome
- ^ http://jhs.sagepub.com/cgi/content/abstract/22/1/128
- ^ Arendt-Nielsen, Lars. "Patients Suffering from Ehlers Danlos Syndrome Type III Do Not Respond to Local Anesthetics". http://www.ednf.org/index.php?option=com_content&task=view&id=1337&Itemid=88889208.
- ^ Lind J, Wallenburg HC (April 2002). "Pregnancy and the Ehlers-Danlos syndrome: a retrospective study in a Dutch population". Acta Obstetricia et Gynecologica Scandinavica 81 (4): 293–300. doi:10.1034/j.1600-0412.2002.810403.x. PMID 11952457.
- ^ http://www.nlm.nih.gov/medlineplus/ency/article/001468.htm
- ^ The Press Enterprise, Redlands mother stung by untrue suspicions presses for accountability in child abuse inquiries, 2008-04-03
- ^ http://rarediseases.about.com/cs/ehlersdanlossynd/a/102603.htm
- ^ http://emedicine.medscape.com/article/943567-treatment
- ^ http://www.medscape.com/viewarticle/466834_3
- ^ http://www.digitalspy.com/soaps/s3/coronation-street/news/a221287/houston-hits-out-at-preconceived-ideas.html
- ^ Yücel, D (January 1995). "Was Paganini born with Ehlers-Danlos syndrome phenotype 4 or 3?". Clin Chem 41 (1): 124–5. PMID 7813066.
- ^ http://www.news.com.au/weird-true-freaky/watch-pianist-evan-petrone-bends-arms-backwards-to-play-clocks-by-coldplay/story-e6frflri-1226130610929
- ^ http://www.youtube.com/watch?v=OGXn6HuCodE&feature=PlayList&p=838DA384930CD363&index=13
- ^ Halper et al. “Degenerative suspensory ligament desmitis as a systemic disorder characterized by proteoglycan accumulation”. Department of Pathology, College of Veterinary Medicine, The University of Georgia, Athens, GA 2006
- ^ Halper et al. “Glycan profiling of a defect in decorin glycosylation in equine systemic proteoglycan accumulation, a potential model of progeroid form of Ehlers-Danlos syndrome”. Department of Pathology, College of Veterinary Medicine, The University of Georgia, Athens, GA 2010
External links
- Ehlers-Danlos Syndrome, Classic Type. Includes: Ehlers-Danlos Syndrome Type I, Ehlers-Danlos Syndrome Type II
- University of Washington Medicine Multi-page explanation of EDS, including symptoms, genetics, diagnosis, and treatment
- National Institute of Health on Hypermobility EDS
- GeneReviews/NCBI/NIH/UW entry on Ehlers-Danlos Syndrome Type IV
- eds at NIH/UW GeneTests
- Joint & Bone - Ehlers-Danlos/Joint Hypermobility Syndrome - Proprioception
- Ehlers–Danlos National Foundation
Genetic disorder, extracellular: scleroprotein disease (excluding laminin and keratin) Collagen disease COL1: Osteogenesis imperfecta · Ehlers–Danlos syndrome, types 1, 2, 7
COL2: Hypochondrogenesis · Achondrogenesis type 2 · Stickler syndrome · Marshall syndrome · Spondyloepiphyseal dysplasia congenita · Spondyloepimetaphyseal dysplasia, Strudwick type · Kniest dysplasia (see also C2/11)
COL3: Ehlers–Danlos syndrome, types 3 & 4 (Sack–Barabas syndrome)
COL4: Alport syndrome
COL5: Ehlers–Danlos syndrome, types 1 & 2
COL6: Bethlem myopathy · Ullrich congenital muscular dystrophy
COL7: Epidermolysis bullosa dystrophica · Recessive dystrophic epidermolysis bullosa · Bart syndrome · Transient bullous dermolysis of the newborn
COL8: Fuchs' dystrophy 1
COL9: Multiple epiphyseal dysplasia 2, 3, 6
COL10: Schmid metaphyseal chondrodysplasia
COL11: Weissenbacher–Zweymüller syndrome · Otospondylomegaepiphyseal dysplasia (see also C2/11)
COL17: Bullous pemphigoidLaminin Junctional epidermolysis bullosa · Laryngoonychocutaneous syndromeOther Congenital stromal corneal dystrophy · Raine syndrome · Urbach–Wiethe disease · TECTA (DFNA8/12, DFNB21)Translation Ribosome: Diamond–Blackfan anemia · FMR1 (Fragile X syndrome, Fragile X-associated tremor/ataxia syndrome, Premature ovarian failure 1)
Initiation factor: Leukoencephalopathy with vanishing white matter
snRNP: Retinitis pigmentosa 33Posttranslational modification E1: X-linked spinal muscular atrophy 2
E3: Johanson–Blizzard syndrome · Von Hippel–Lindau disease · 3-M syndrome · Angelman syndrome
Deubiquitinating enzyme: Machado–Joseph disease · Aneurysmal bone cyst · Multiple familial trichoepithelioma 1Othersee also genetic translation, posttranslational modification
B structural (perx, skel, cili, mito, nucl, sclr) · DNA/RNA/protein synthesis (drep, trfc, tscr, tltn) · membrane (icha, slcr, atpa, abct, othr) · transduction (iter, csrc, itra), trfkCategories:- Abnormalities of dermal fibrous and elastic tissue
- Congenital disorders
- Connective tissue diseases
- Contortion
- Diseases involving the fasciae
- Syndromes
- Collagen disease



Wikimedia Foundation. 2010.