- Marfan syndrome
-
Marfan syndrome Classification and external resources 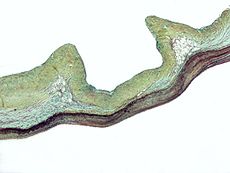
Micrograph demonstrating myxomatous degeneration of the aortic valve, a common manifestation of Marfan syndromeICD-10 Q87.4 ICD-9 759.82 OMIM 154700 DiseasesDB 7845 MedlinePlus 000418 eMedicine ped/1372 orthoped/414 MeSH D008382 Marfan syndrome (also called Marfan's syndrome) is a genetic disorder of the connective tissue. People with Marfan's tend to be unusually tall, with long limbs and long, thin fingers.
It is inherited as a dominant trait. It is carried by a gene called FBN1, which encodes a connective protein called fibrillin-1.[1][2] People have a pair of FBN1 genes. Because it is dominant, people who have inherited one affected FBN1 gene from either parent will have Marfan's.
Marfan syndrome has a range of expressions, from mild to severe. The most serious complications are defects of the heart valves and aorta. It may also affect the lungs, eyes, the dural sac surrounding the spinal cord, skeleton and the hard palate.
In addition to being a connective protein that forms the structural support for tissues outside the cell, the normal fibrillin-1 protein binds to another protein, transforming growth factor beta (TGF-β).[2] TGF-β has deleterious effects on vascular smooth muscle development and the integrity of the extracellular matrix. Researchers now believe that secondary to mutated fibrillin there is excessive TGF-β at the lungs, heart valves, and aorta, and this weakens the tissues and causes the features of Marfan syndrome.[3] Since angiotensin II receptor blockers (ARBs) also reduce TGF-β, they have tested this by giving ARBs (losartan, etc.) to a small sample of young, severely affected Marfan syndrome patients. In some patients, the growth of the aorta was indeed reduced.[4]
Marfan syndrome is named after Antoine Marfan,[5] the French pediatrician who first described the condition in 1896.[6][7] The gene linked to the disease was first identified by Francesco Ramirez in 1991.[8]
Contents
Signs and symptoms
Although there are some unique signs or symptoms of Marfan syndrome, the constellation of long limbs, the dislocated lenses and the aortic root dilation are generally sufficient to make the diagnosis with reasonable confidence. There are more than 30 other clinical features that are variably associated with the syndrome, most involving the skeleton, skin, and joints. There may be considerable clinical variability within families that carry the identical mutation.
Skeletal system
Most of the readily visible signs are associated with the skeletal system. Many individuals with Marfan syndrome grow to above average height. Some have long slender limbs with long fingers and toes (arachnodactyly). This condition of elongated limbs is known as dolichostenomelia. An individual's arms may be disproportionately long, with thin, weak wrists. In addition to affecting height and limb proportions, Marfan syndrome can produce other skeletal anomalies. Abnormal curvature of the spine (scoliosis) is not uncommon. Abnormal indentation (pectus excavatum) or protrusion (pectus carinatum) of the sternum. Other signs include abnormal joint flexibility, a high palate, malocclusions, flat feet, hammer toes, stooped shoulders, unexplained stretch marks on the skin. It can also cause pain in the joints, bones and muscles in some patients. Some people with Marfan have speech disorders resulting from symptomatic high palates and small jaws. Early osteoarthritis may occur.
Eyes
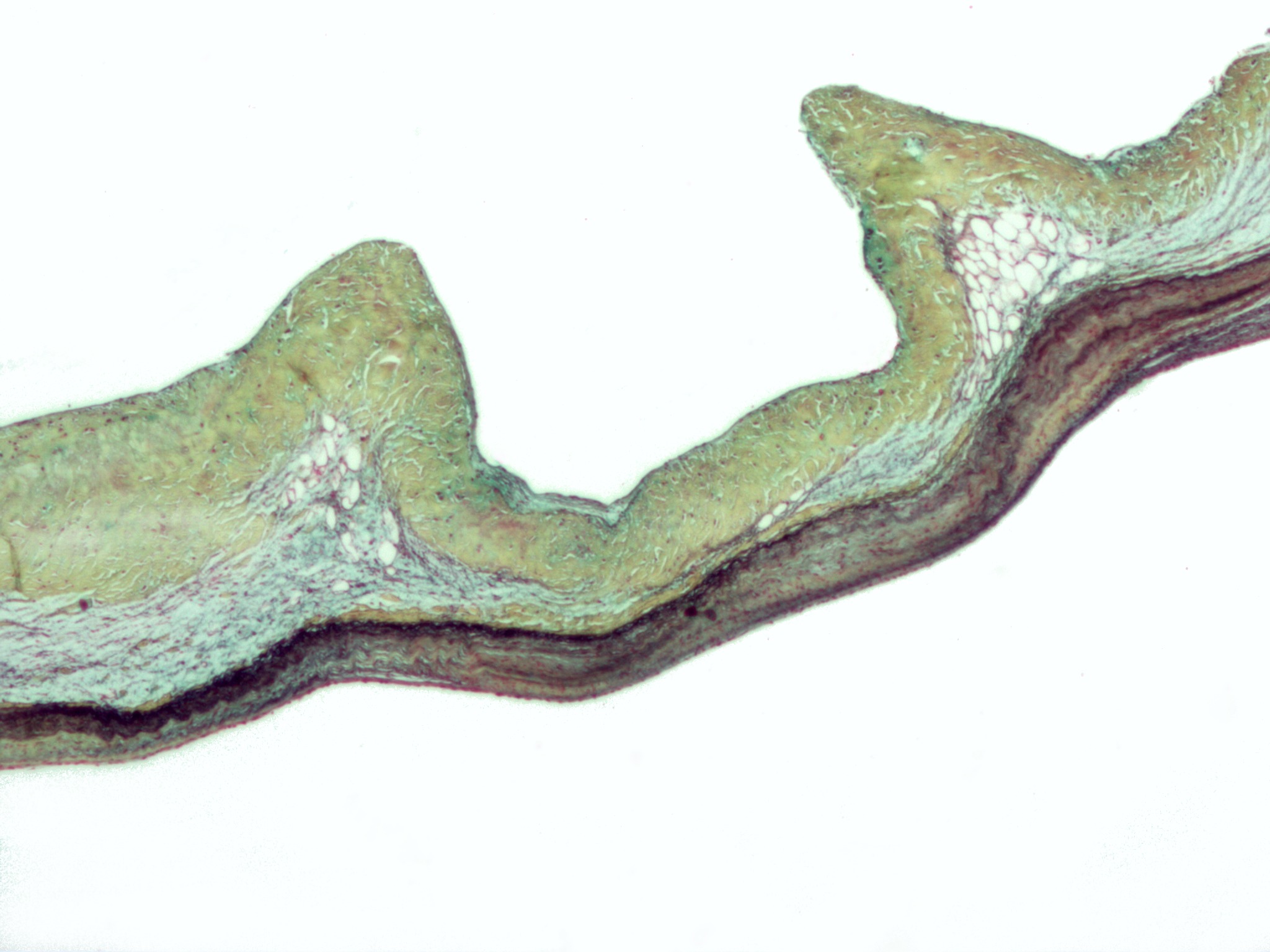
 Lens dislocation in Marfan's syndrome; the lens was kidney-shaped and was resting against the ciliary body.
Lens dislocation in Marfan's syndrome; the lens was kidney-shaped and was resting against the ciliary body.
Marfan syndrome can also seriously affect the eyes and vision. Nearsightedness and astigmatism are common, but farsightedness can also result. Subluxation (dislocation) of the crystalline lens in one or both eyes (ectopia lentis) (in 80% of patients) also occurs and may be detected by an ophthalmologist or optometrist using a slit-lamp biomicroscope. In Marfan's the dislocation is typically superotemporal whereas in the similar condition homocystinuria, the dislocation is inferonasal. Sometimes eye problems appear only after the weakening of connective tissue has caused detachment of the retina.[9] Early onset glaucoma can be another related problem.
Cardiovascular system
The most serious signs and symptoms associated with Marfan syndrome involve the cardiovascular system. Undue fatigue, shortness of breath, heart palpitations, racing heartbeats, or Angina pectoris with pain radiating to the back, shoulder, or arm. Cold arms, hands and feet can also be linked to Marfan's syndrome because of inadequate circulation. A heart murmur, abnormal reading on an ECG, or symptoms of angina can indicate further investigation. The signs of regurgitation from prolapse of the mitral or aortic valves (which control the flow of blood through the heart) result from cystic medial degeneration of the valves, which is commonly associated with Marfan's syndrome (see mitral valve prolapse, aortic regurgitation). However, the major sign that would lead a doctor to consider an underlying condition is a dilated aorta or an aortic aneurysm. Sometimes, no heart problems are apparent until the weakening of the connective tissue (cystic medial degeneration) in the ascending aorta causes an aortic aneurysm or aortic dissection, a surgical emergency. An aortic dissection is most often fatal and presents with pain radiating down the back, giving a tearing sensation.
Because of the underlying connective tissue abnormalities that cause Marfan syndrome, there is an increased incidence of dehiscence of prosthetic mitral valve.[10] Care should be taken to attempt repair of damaged heart valves rather than replacement.
During pregnancy, even in the absence of preconceived cardiovascular abnormality, women with Marfan syndrome are at significant risk of aortic dissection, which is often fatal even when rapidly treated. For this reason, women with Marfan syndrome should receive a thorough assessment prior to conception, and echocardiography should be performed every six to ten weeks during pregnancy, to assess the aortic root diameter. For most women, safe vaginal delivery is possible.[11]
Lungs
Marfan syndrome is a risk factor for spontaneous pneumothorax. In spontaneous unilateral pneumothorax, air escapes from a lung and occupies the pleural space between the chest wall and a lung. The lung becomes partially compressed or collapsed. This can cause pain, shortness of breath, cyanosis, and, if not treated, death. Marfan syndrome has also been associated with sleep apnea and idiopathic obstructive lung disease.
Central nervous system
Another condition that can reduce the quality of life for an individual, though not life-threatening, is dural ectasia, the weakening of the connective tissue of the dural sac, the membrane that encases the spinal cord. Dural ectasia can be present for a long time without producing any noticeable symptoms. Symptoms that can occur are lower back pain, leg pain, abdominal pain, other neurological symptoms in the lower extremities, or headaches. Such symptoms usually diminish when the individual lies flat on his or her back. These types of symptoms might lead a doctor to order an X-ray of the lower spine. Dural ectasia is usually not visible on an X-ray in the early phases. A worsening of symptoms and the lack of finding any other cause should eventually lead a doctor to order an upright MRI of the lower spine. Dural ectasia that has progressed to the point of causing these symptoms would appear in an upright MRI image as a dilated pouch that is wearing away at the lumbar vertebrae.[9] Other spinal issues associated with Marfan include degenerative disk disease and spinal cysts. Marfan syndrome is also associated with dysautonomia.
Pathogenesis
Marfan syndrome is caused by mutations in the FBN1 gene on chromosome 15,[12] which encodes a glycoprotein called fibrillin-1, a component of the extracellular matrix. The Fibrillin 1 protein is essential for the proper formation of the extracellular matrix including the biogenesis and maintenance of elastic fibers. The extracellular matrix is critical for both the structural integrity of connective tissue but also serves as a reservoir for growth factors.[13] Elastin fibers are found throughout the body but are particularly abundant in the aorta, ligaments and the ciliary zonules of the eye; consequently, these areas are among the worst affected.
A transgenic mouse has been created carrying a single copy of a mutant fibrillin 1, a mutation similar to that found in the human fibrillin 1 gene that is known to cause Marfan syndrome. This mouse strain recapitulates many of the features of the human disease and promises to provide insights into the pathogenesis of the disease. Reducing the level of normal fibrillin-1 causes a Marfan-related disease in mice.[14]
Transforming growth factor beta (TGFβ) plays an important role in Marfan syndrome. Fibrillin-1 directly binds a latent form of TGFβ keeping it sequestered and unable to exert its biological activity. The simplest model of Marfan syndrome suggests that reduced levels of fibrillin-1 allow TGFβ levels to rise due to inadequate sequestration. Although it is not proven how elevated TGFβ levels are responsible for the specific pathology seen with the disease, an inflammatory reaction releasing proteases that slowly degrade the elastin fibers and other components of the extracellular matrix is known to occur. The importance of the TGFβ pathway was confirmed with the discovery of a similar syndrome Loeys-Dietz syndrome involving the TGFβR2 gene on chromosome 3, a receptor protein of TGFβ.[15] Marfan syndrome has often been confused with Loeys-Dietz syndrome, because of the considerable clinical overlap between the two pathologies.[16]
Diagnosis
Diagnostic criteria of Marfan syndrome were agreed internationally in 1996.[17] A diagnosis of Marfan syndrome is based on family history and a combination of major and minor indicators of the disorder, rare in the general population, that occur in one individual. For example: four skeletal signs with one or more signs in another body system such as ocular and cardiovascular in one individual. The following conditions may result from Marfan syndrome but may also occur in people without any known underlying disorder.
Revised Ghent Nosology
According to the US National Marfan Foundation, in 2010 the Ghent Nosology was revised and new diagnostic criteria superseded the previous agreement made in 1996. There are seven new criteria that can lead to a diagnosis:[unreliable medical source?][27]
- Aortic root dilatation (defined as a Z-Score of 2 or higher) and ectopia lentis (dislocated optic lens)
- Aortic root dilatation and a confirmed mutation in the Fibrilin-1 gene (FBN1)
- Aortic root dilatation and a systemic Score of 7 or higher (see systemic score below)
- Ectopia lentis and a FBN1 mutation known to cause an aortic aneurysm
- Ectopia lentis and a family history with someone meeting the above criteria
- Systemic score of 7 or greater and a family history of Marfan syndrome
- Aortic root dilatation (Z-Score of 2 or higher for 20 years old or older, 3 or higher for people under the age of 20) and a family history of Marfan syndrome
Differential diagnosis
The following disorders have similar signs and symptoms of Marfan syndrome:
- Congenital Contractural Arachnodactyly (CCA) or Beals Syndrome
- Ehlers–Danlos syndrome
- Homocystinuria
- Loeys–Dietz syndrome
- MASS phenotype
- Shprintzen–Goldberg syndrome[28]
- Stickler syndrome
- Multiple endocrine neoplasia, type 2B
Management
There is no cure for Marfan syndrome, but life expectancy has increased significantly over the last few decades, and clinical trials are underway for a promising new treatment.[29] At present (2011), the syndrome is treated by simply addressing each issue as it arises and, in particular, preventive medication even for young children to slow progression of aortic dilation if such exists.
Marfan's syndrome is passed on to offspring dominantly. This means that a child with one parent a bearer of the gene have a 50% probability of getting the syndrome. However, as the gene causing Marfan's syndrome is known, there in principle are arduous genetic techniques of circumventing this. In 1996 the first preimplantation genetic testing therapy for Marfan's was conducted,[30] in essence PGT means conducting a genetic testing on early stage IVF embryo cells and discarding those embryos affected by the Marfan mutation.
Regular checkups by a cardiologist are needed to monitor the health of the heart valves and the aorta. The goal of treatment is to slow the progression of aortic dilation and damage to heart valves by eliminating arrythmias, minimizing the heart rate, and minimizing blood pressure. Beta blockers have been used to control arrythmias and slow the heart rate. Other medications might be needed to further minimize blood pressure without slowing the heart rate, such as ACE inhibitors and angiotensin II receptor antagonists, also ]known as angiontensin receptor blockers (ARBs). If the dilation of the aorta progresses to a significant diameter aneurysm, causes a dissection or a rupture, or leads to failure of the aortic or other valve, then surgery (possibly a composite aortic valve graft [CAVG] or valve-sparing procedure) becomes necessary. Although aortic graft surgery (or any vascular surgery) is a serious undertaking it is generally successful if undertaken on an elective basis.[31] Surgery in the setting of acute aortic dissection or rupture is considerably more problematic. Elective aortic valve/graft surgery is usually considered when aortic root diameter reaches 50 millimeters (2.0 inches), but each case needs to be specifically evaluated by a qualified cardiologist. New valve-sparing surgical techniques are becoming more common.[32] As Marfan patients live longer, other vascular repairs are becoming more common, e.g., repairs of descending thoractic aortic aneurysms and aneurysms of vessels other than the aorta.
The skeletal and ocular manifestations of Marfan syndrome can also be serious, although not life-threatening. These symptoms are usually treated in the typical manner for the appropriate condition, such as with various kinds of pain medication or muscle relaxants. It is also common for patients to receive treatment from a physiotherapist, using TENS therapy, ultrasound and skeletal adjustment.[citation needed] This can also affect height, arm length, and life span. A physiotherapist can also help improve function and prevent injuries in individuals with Marfan's. The Nuss procedure is now being offered to people with Marfan syndrome to correct 'sunken chest' or (pectus excavatum).[33] Because Marfan may cause spinal abnormalities that are asymptomatic, any spinal surgery contemplated on a Marfan patient should only follow detailed imaging and careful surgical planning, regardless of the indication for surgery.
Treatment of a spontaneous pneumothorax is dependent on the volume of air in the pleural space and the natural progression of the individual's condition. A small pneumothorax might resolve without active treatment in one to two weeks. Recurrent pneumothoraces might require chest surgery. Moderately sized pneumothoraces might need chest drain management for several days in a hospital. Large pneumothoraces are likely to be medical emergencies requiring emergency decompression.
Research in laboratory mice has suggested that the angiotensin II receptor antagonist losartan, which appears to block TGF-beta activity, can slow or halt the formation of aortic aneurysms in Marfan syndrome.[34] A large clinical trial sponsored by the National Institutes of Health comparing the effects of losartan and atenolol on the aortas of Marfan patients was scheduled to begin in early 2007, coordinated by Johns Hopkins.[35]
Epidemiology
Marfan syndrome affects males and females equally,[36] and the mutation shows no ethnic or geographical bias.[37] Estimates indicate that approximately 1 in 3,000 to 5,000 individuals have Marfan syndrome.[37] Each parent with the condition has a 50% risk of passing the genetic defect on to any child due to its autosomal dominant nature. Most individuals with Marfan syndrome have another affected family member - approximately 15–30% of all cases are due to de novo genetic mutations[13]—such spontaneous mutations occur in about 1 in 20,000 births. Marfan syndrome is also an example of dominant negative mutation and haploinsufficiency.[38][39] It is associated with variable expressivity; incomplete penetrance has not been definitively documented.
History
Marfan syndrome is named after Antoine Marfan,[5] the French pediatrician who first described the condition in 1896 after noticing striking features in a 5-year-old girl.[6][7] The gene linked to the disease was first identified by Francesco Ramirez at the Mount Sinai Medical Center in New York City in 1991.[8]
Musicians and composers Niccolò Paganini,[40] Sergei Rachmaninoff,[41] and Robert Johnson[42] are thought to have had the disease. Abraham Lincoln may or may not have had Marfan's syndrome, although he undoubtedly had some of the normal characteristic features.[43][44][45] According to a 2007 theory, it is perhaps more likely that he had a different disorder, multiple endocrine neoplasia type 2B, that caused skeletal features almost identical to Marfan syndrome.[46]
Society and culture
Contributors to public perception of Marfan syndrome include Flo Hyman, an Olympic silver medalist in Women's Volleyball (1984) who died suddenly at a match from an aortic dissection;[47] Jonathan Larson, the author and composer of Rent, who died from an aortic dissection the day before the off-Broadway opening of Rent;[48] and Vincent Schiavelli, an actor and spokesperson for the National Marfan Foundation.[49]
References
- ^ Kainulainen K, Karttunen L, Puhakka L, Sakai L, Peltonen L (January 1994). "Mutations in the fibrillin gene responsible for dominant ectopia lentis and neonatal Marfan syndrome". Nat. Genet. 6 (1): 64–9. doi:10.1038/ng0194-64. PMID 8136837.
- ^ a b Dietz HC, Loeys B, Carta L, Ramirez F (2005). "Recent progress towards a molecular understanding of Marfan syndrome". Am J Med Genet C Semin Med Genet 139: 4–9.
- ^ Robbins and Cotran Pathological Basis of Disease, Kumar et al; 8th Edition, Saunders Elsevier Publishing, 2010
- ^ Pyeritz RE (June 2008). "A small molecule for a large disease". N. Engl. J. Med. 358 (26): 2829–31. doi:10.1056/NEJMe0804008. PMID 18579819.
- ^ a b (French) Marfan, Antoine (1896). "Un cas de déformation congénitale des quartre membres, plus prononcée aux extrémitiés, caractérisée par l'allongement des os avec un certain degré d'amincissement [A case of congenital deformation of the four limbs, more pronounced at the extremities, characterized by elongation of the bones with some degree of thinning]". Bulletins et memoires de la Société medicale des hôspitaux de Paris 13 (3rd series): 220–226.
- ^ a b Johns Hopkins Comprehensive Marfan Center. John Hopkins Medicine. Retrieved on January 6, 2009.
- ^ a b Antoine Bernard-Jean Marfan at Who Named It?
- ^ a b Brown P (July 27, 1991). "Marfan syndrome linked to gene". New Scientist. Retrieved on August 11, 2008.
- ^ a b "Marfan Syndrome". Mayo Clinic. http://www.mayoclinic.com/health/marfan-syndrome/DS00540/DSECTION=2. Retrieved January 12, 2007.
- ^ Braunwald's Heart Disease ~ A Textbook of Cardiovascular Medicine, Seventh Edition. United States of America: Elseview Saunders. 2005. pp. 1894. ISBN 0-7216-0509-5.
- ^ Chen H (June 4, 2007). "Marfan Syndrome". eMedicine. http://emedicine.medscape.com/article/946315-print. Retrieved June 25, 2007.
- ^ McKusick V (1991). "The defect in Marfan syndrome". Nature 352 (6333): 279–81. Bibcode 1991Natur.352..279M. doi:10.1038/352279a0. PMID 1852198.
- ^ a b Cotran; Kumar, Collins (1998). Robbins Pathologic Basis of Disease. Philadelphia: W.B Saunders Company. ISBN 0-7216-7335-X.
- ^ Pereira L, Lee SY, Gayraud B, et al. (March 1999). "Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1". Proceedings of the National Academy of Sciences of the United States of America 96 (7): 3819–23. Bibcode 1999PNAS...96.3819P. doi:10.1073/pnas.96.7.3819. PMC 22378. PMID 10097121. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=22378.
- ^ Entrez Gene (2007). "TGFBR2 transforming growth factor, beta receptor II" (Entrez gene entry). NCBI. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=gene&dopt=full_report&list_uids=7048. Retrieved January 11, 2007.
- ^ "Related Disorders: Loeys-Dietz". National Marfan Foundation. Archived from the original on September 25, 2006. http://web.archive.org/web/20060925143304/http://www.marfan.org/nmf/GetContentRequestHandler.do?menu_item_id=84. Retrieved January 11, 2007.
- ^ De Paepe A, Devereux RB, Dietz HC, Hennekam RC, Pyeritz RE (April 1996). "Revised diagnostic criteria for the Marfan syndrome". Am. J. Med. Genet. 62 (4): 417–26. doi:10.1002/(SICI)1096-8628(19960424)62:4<417::AID-AJMG15>3.0.CO;2-R. PMID 8723076.
- ^ Finkbohner R, Johnston D, Crawford ES, Coselli J, Milewicz DM (February 1995). "Marfan syndrome. Long-term survival and complications after aortic aneurysm repair". Circulation 91 (3): 728–33. PMID 7828300. http://circ.ahajournals.org/cgi/content/full/91/3/728.
- ^ "Marfan Syndrome - Signs and Symptoms". www.ucsfhealth.org. http://www.ucsfhealth.org/adult/medical_services/heart_care/marfan/conditions/marfan/signs.html. Retrieved 2009-08-28.
- ^ "The Marfan Trust - What is Marfan Syndrome?". www.marfantrust.org. http://www.marfantrust.org/marfan-syndrome.html. Retrieved 2009-08-28.[dead link]
- ^ "Marfan Syndrome: The Similarities to Copper Deficiency". www.ctds.info. http://www.ctds.info/marfan_syndrome.html. Retrieved 2009-08-29.
- ^ a b c "Marfan syndrome: MedlinePlus Medical Encyclopedia". www.nlm.nih.gov. http://www.nlm.nih.gov/medlineplus/ency/article/000418.htm. Retrieved 2009-08-28.
- ^ "Marfan syndrome - Genetics Home Reference". ghr.nlm.nih.gov. http://ghr.nlm.nih.gov/condition%3Dmarfansyndrome. Retrieved 2009-08-28.
- ^ Kohlmeier L, Gasner C, Bachrach LK, Marcus R (October 1995). "The bone mineral status of patients with Marfan syndrome". Journal of Bone and Mineral Research 10 (10): 1550–5. doi:10.1002/jbmr.5650101017. PMID 8686512.
- ^ a b "About Marfan Syndrome: Features". National Marfan Foundation. http://www.marfan.org/marfan/2320/Features#Other. Retrieved 2009-08-28.
- ^ "Living with Marfan Syndrome: Dental issues". National Marfan Foundation. http://www.marfan.org/marfan/2711/Dental-Issues#TMJ. Retrieved 2009-08-28.
- ^[unreliable medical source?]"2010 Revised Ghent Nosology". National Marfan Foundation. http://www.marfan.org/marfan/4265/2010-Revised-Ghent-Nosology. Retrieved 2011-01-31.
- ^ "Shprintzen–Goldberg Syndrome – GeneReviews – NCBI Bookshelf". http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=sgs. Retrieved 2009-08-29.
- ^ Freeman, Elaine (Fall 2007). "A Silver Bullet for Blake". Johns Hopkins Magazine. http://www.hopkinsmedicine.org/hmn/F07/feature1.cfm.
- ^ http://www.ncbi.nlm.nih.gov/pubmed/9239687
- ^ "Elective Aortic Root Surgery in Marfan Syndrome Appears Safe and Durable: Presented at STS" (Press release). Doctor's Guide. January 31, 2008. http://www.docguide.com/news/content.nsf/news/852571020057CCF6852573E1005A0BDC. Retrieved January 13, 2009.
See also:- Cameron DE, Vricella LA (2005). "Valve-sparing aortic root replacement in Marfan syndrome". Seminars in Thoracic and Cardiovascular Surgery 8 (1): 103–11. doi:10.1053/j.pcsu.2005.03.001. PMID 15818365.
- Gott VL, Cameron DE, Alejo DE, et al. (February 2002). "Aortic root replacement in 271 Marfan patients: a 24-year experience". The Annals of Thoracic Surgery 73 (2): 438–43. doi:10.1016/S0003-4975(01)03336-7. PMID 11845856.
- Bethea BT, Fitton TP, Alejo DE, et al. (September 2004). "Results of aortic valve-sparing operations: experience with remodeling and reimplantation procedures in 65 patients". The Annals of Thoracic Surgery 78 (3): 767–72; discussion 767–72. doi:10.1016/j.athoracsur.2004.03.040. PMID 15336989.
- ^ "Heart Surgery for Marfan Syndrome". Mayo Clinic. Archived from the original on December 18, 2006. http://web.archive.org/web/20061218031510/http://www.mayoclinic.org/marfan-syndrome/heartsurgery.html. Retrieved January 12, 2007.
- ^ "Overview of the Nuss Procedure for Pectus Excavatum". Children's Hospital of The King's Daughters. http://www.chkd.org/services/nussprocedure/Overview.aspx. Retrieved January 12, 2007.
- ^ Habashi JP, Judge DP, Holm TM, et al. (April 2006). "Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome". Science 312 (5770): 117–21. Bibcode 2006Sci...312..117H. doi:10.1126/science.1124287. PMC 1482474. PMID 16601194. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1482474.
- ^ "Atenolol vs. Losartan in Individuals with Marfan Syndrome Clinial Trial". National Marfan Foundation. Archived from the original on September 25, 2006. http://web.archive.org/web/20060925151335/http://www.marfan.org/nmf/GetSubContentRequestHandler.do?sub_menu_item_content_id=147&menu_item_id=91. Retrieved January 12, 2007.
- ^ Fusar-Poli P, Klersy C, Stramesi F, Callegari A, Arbustini E, Politi P (2008). "Determinants of quality of life in Marfan syndrome". Psychosomatics 49 (3): 243–8. doi:10.1176/appi.psy.49.3.243. PMID 18448780. http://psy.psychiatryonline.org/cgi/content/full/49/3/243.
- ^ a b Keane MG, Pyeritz RE (May 2008). "Medical management of Marfan syndrome". Circulation 117 (21): 2802–13. doi:10.1161/CIRCULATIONAHA.107.693523. PMID 18506019. http://circ.ahajournals.org/cgi/content/full/117/21/2802.
- ^ Judge DP, Biery NJ, Keene DR, et al. (July 2004). "Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome". The Journal of Clinical Investigation 114 (2): 172–81. doi:10.1172/JCI20641. PMC 449744. PMID 15254584. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=449744.
- ^ Judge DP, Dietz HC (December 2005). "Marfan's syndrome". Lancet 366 (9501): 1965–76. doi:10.1016/S0140-6736(05)67789-6. PMC 1513064. PMID 16325700. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1513064.
- ^ From Paganini stories myths. The AFU and Urban Legends Archive. Retrieved on January 13, 2009; based primarily on Schoenfeld MR (January 1978). "Nicolo Paganini. Musical magician and Marfan mutant?". JAMA 239 (1): 40–2. doi:10.1001/jama.239.1.40. PMID 336919. http://jama.ama-assn.org/cgi/content/abstract/239/1/40.
- ^ Wolf P (November 2001). "Creativity and chronic disease. Sergei Rachmaninov (1873-1943)". West. J. Med. 175 (5): 354. doi:10.1136/ewjm.175.5.354. PMC 1071626. PMID 11694497. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1071626.
- ^ Connel D (September 2, 2006). "Retrospective blues: Robert Johnson—an open letter to Eric Clapton". British Medical Journal 333 (7566): 489. doi:10.1136/bmj.333.7566.489. http://www.bmj.com/cgi/content/full/333/7566/489.
- ^ Marfan syndrome. MayoClinic.com. Retrieved on January 6, 2009.
- ^ Aneurysm and dissection of the aorta. NIH. Retrieved on January 6, 2009.
- ^ Swartz, Harold (May 1972), "Abraham Lincoln and Aortic Insufficiency: The Declining Health of the President", California Medical, PMC 1518411, http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1518411
- ^ From The Last - and Greatest - Lincoln Mystery. The Physical Lincoln. Retrieved on January 13, 2009; reporting on Sotos JG (2008). The Physical Lincoln. Mt. Vernon Book Systems. ISBN 9780981819327
- ^ "Flo Hyman". Volleyball Hall of Fame. Archived from the original on January 30, 2008. http://web.archive.org/web/20080130003123/http://www.volleyhall.org/hyman.html. Retrieved January 6, 2009.
- ^ Lawrence Van Gelder (1996-12-13). "On the Eve of a New Life, an Untimely Death". The New York Times. http://query.nytimes.com/gst/fullpage.html?res=9C07E0DC143EF930A25751C1A960958260. Retrieved 2008-07-17.
- ^ "NMF Mourns the Loss of its Honorary Co-Chair, Vincent Schiavelli". National Marfan Foundation. http://www.marfan.org/marfan/2635/APR--NMF-Mourns-Vincent-Schiavelli. Retrieved April 10, 2011.
External links
- Marfan syndrome at the Open Directory Project
- Marfan Syndrome Diagnosis (www.nature.com)
- Marfan syndrome - NIH's Office of Rare Diseases
- GeneReviews/NCBI/UW/NIH entry on Marfan syndrome
- GeneReviews/NCBI/NIH/UW entry on Shprintzen-Goldberg Syndrome
- National Marfan Foundation
- Marfan Trust UK
- Canadian Marfan Association
Congenital abnormality · multiple abnormalities (Q87, 759.7) Craniofacial Short stature 1q21.1 deletion syndrome · Aarskog–Scott syndrome · Cockayne syndrome · Cornelia de Lange Syndrome · Dubowitz syndrome · Noonan syndrome · Robinow syndrome · Silver–Russell syndrome · Seckel syndrome · Smith-Lemli-Opitz syndrome-Turner syndromeLimbs Overgrowth Laurence-Moon-Bardet-Biedl Combined/other,
known locus3 (Zimmerman-Laband syndrome) · 4/13 (Fraser syndrome) · 8 (Branchio-oto-renal syndrome) · 12 (Keutel syndrome, Timothy syndrome) · 15 (Marfan syndrome) · 19 (Donohue syndrome)Systemic CT disorders (M32–M36, 710) General Other hypersensitivity/autoimmune Other Categories:- Abnormalities of dermal fibrous and elastic tissue
- Diseases involving the fasciae
- Systemic connective tissue disorders
- Syndromes
- Cytoskeletal defects
Wikimedia Foundation. 2010.