- Sotos syndrome
-
Sotos syndrome Classification and external resources 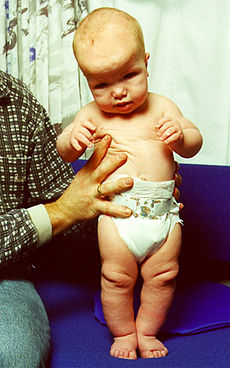
ICD-10 Q87.3 ICD-9 759.89 OMIM 117550 DiseasesDB 29134 Sotos syndrome (cerebral gigantism) is a rare genetic disorder characterized by excessive physical growth during the first 2 to 3 years of life. The disorder may be accompanied by mild mental retardation, delayed motor, cognitive, and social development, hypotonia (low muscle tone), and speech impairments. Children with Sotos syndrome tend to be large at birth and are often taller, heavier, and have larger heads (macrocephaly) than is normal for their age. Signs of the disorder, which vary among individuals, include a disportionately large and long head with a slightly protrusive forehead, large hands and feet, hypertelorism (an abnormally increased distance between the eyes), and downslanting eyes. Clumsiness, an awkward gait, and unusual aggressiveness or irritability may also occur. Although most cases of Sotos syndrome occur sporadically, familial cases have also been reported. It is similar to Weaver syndrome.
Contents
Epidemiology
Incidence is approximately 1 in 14,000 births.
Signs and symptoms
Characterized by overgrowth and advanced bone age. Affected individuals are dysmorphic with macrodolichocephaly, downslanting palpebral fissures and a pointed chin. The facial appearance is most notable in early childhood. Affected infants and children tend to grow quickly; they are significantly taller than their siblings and peers and have an unusually large head. Adult height is usually in the normal range, however.
People with Sotos syndrome often have intellectual impairment, and most also have behavioral problems. Frequent behavioral issues include attention deficit hyperactivity disorder (ADHD), phobias, obsessions and compulsions, tantrums, and impulsive behaviors. Problems with speech and language are also common. Affected individuals often have problems with sound production, stuttering, and a monotone voice. Additionally, weak muscle tone (hypotonia) may delay other aspects of early development, particularly motor skills such as sitting and crawling.
Other signs of Sotos syndrome include an abnormal side-to-side curvature of the spine (scoliosis), seizures, heart or kidney defects, hearing loss, and problems with vision. Some infants with this disorder experience yellowing of the skin and whites of the eyes (jaundice) and poor feeding. A few people with Sotos syndrome have developed cancer, most often in childhood, but no single form of cancer has been associated with this condition. It remains uncertain whether Sotos syndrome increases the risk of specific types of cancer. If people with this disorder have any increased cancer risk, their risk is only slightly greater than that of the general population.
Genetics
Mutations in the NSD1 gene cause Sotos syndrome.[1] The NSD1 gene provides instructions for making a protein that is involved in normal growth and development. The function of this protein is unknown, however. In the Japanese population, the most common genetic change leading to Sotos syndrome deletes genetic material from the region of chromosome 5 containing the NSD1 gene. In other populations, small mutations within the NSD1 gene occur more frequently. Genetic changes involving the NSD1 gene prevent one copy of the gene from producing any functional protein. It is unclear how a reduced amount of this protein during development leads to learning disabilities, overgrowth, and the other features of Sotos syndrome.
About 95 percent of Sotos syndrome cases occur in people with no history of the disorder in their family. Most of these cases result from new mutations involving the NSD1 gene. A few families have been described with more than one affected family member. These cases helped researchers determine that Sotos syndrome has an autosomal dominant pattern of inheritance. Autosomal dominant inheritance means one copy of the altered gene in each cell is sufficient to cause the disorder.
Treatment
There is no standard course of treatment for Sotos syndrome. Treatment is symptomatic.
Prognosis
Sotos syndrome is not a life-threatening disorder and patients may have a normal life expectancy. The initial abnormalities of Sotos syndrome usually resolve as the growth rate becomes normal after the first few years of life. Developmental delays may improve in the school-age years; however, coordination problems may persist into adulthood.
See also
- Perlman syndrome
- Beckwith-Wiedemann syndrome
References
- ^ Melchior L, Schwartz M, Duno M (March 2005). "dHPLC screening of the NSD1 gene identifies nine novel mutations--summary of the first 100 Sotos syndrome mutations". Ann. Hum. Genet. 69 (Pt 2): 222–6. doi:10.1046/j.1529-8817.2004.00150.x. PMID 15720303. http://www.blackwell-synergy.com/openurl?genre=article&sid=nlm:pubmed&issn=0003-4800&date=2005&volume=69&issue=Pt%202&spage=222.
External links
- Information related to orphan diseases and orphan drugs - site is in French
- sotos at NIH/UW GeneTests
Congenital abnormality · multiple abnormalities (Q87, 759.7) Craniofacial Short stature 1q21.1 deletion syndrome · Aarskog–Scott syndrome · Cockayne syndrome · Cornelia de Lange Syndrome · Dubowitz syndrome · Noonan syndrome · Robinow syndrome · Silver–Russell syndrome · Seckel syndrome · Smith-Lemli-Opitz syndrome-Turner syndromeLimbs Overgrowth Laurence-Moon-Bardet-Biedl Bardet–Biedl syndrome · Laurence-Moon syndromeCombined/other,
known locus3 (Zimmerman-Laband syndrome) · 4/13 (Fraser syndrome) · 8 (Branchio-oto-renal syndrome) · 12 (Keutel syndrome, Timothy syndrome) · 15 (Marfan syndrome) · 19 (Donohue syndrome)Categories:- Syndromes
Wikimedia Foundation. 2010.