- Charcot–Marie–Tooth disease
-
Charcot-Marie-Tooth disease Classification and external resources 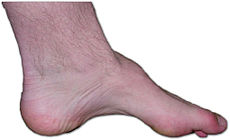
The foot of a person with Charcot-Marie-Tooth. The lack of muscle, a high arch, and claw toes are signs of the genetic disease.ICD-10 G60.0 ICD-9 356.1 DiseasesDB 5815 2343 MedlinePlus 000727 eMedicine orthoped/43 pmr/29 MeSH D002607 Charcot–Marie–Tooth disease- (CMT), known also as Morbus Charcot-Marie-Tooth, Charcot-Marie-Tooth neuropathy, hereditary motor and sensory neuropathy (HMSN), hereditary sensorimotor neuropathy (HSMN), or peroneal muscular atrophy, is an inherited disorder of nerves (neuropathy) that takes different forms. It is characterized by loss of muscle tissue and touch sensation, predominantly in the feet and legs but also in the hands and arms in the advanced stages of disease. Currently incurable, this disease is one of the most common inherited neurological disorders. [1]
Estimates of incidence vary widely from 1 in 380,000 people affected [1] to 1 in 2,500 people affected [1]. This larger figure might equate to approximately 23,000 people in the UK and 125,000 people in the USA.
Contents
Molecular causes
Charcot–Marie–Tooth disease is caused by mutations that cause defects in neuronal proteins. Nerve signals are conducted by an axon with a myelin sheath wrapped around it. Most mutations in CMT affect the myelin sheath. Some affect the axon.
The most common cause of CMT (70-80% of the cases) is the duplication of a large region in chromosome 17p12 that includes the gene PMP22. Some mutations affect the gene MFN2, which codes for a mitochondrial protein. Cells contain separate sets of genes in their nucleus and in their mitochondria. In nerve cells, the mitochondria travel down the long axons. In some forms of CMT, mutated MFN2 causes the mitochondria to form large clusters, or clots, which are unable to travel down the axon towards the synapses. This prevents the synapses from functioning.[2]
CMT is divided into the primary demyelinating neuropathies (CMT1, CMT3, and CMT4) and the primary axonal neuropathies (CMT2), with frequent overlap. Another cell involved in CMT is the Schwann cell, which creates the myelin sheath, by wrapping its plasma membrane around the axon in a structure that is sometimes compared to a Swiss roll.[3]
Neurons, Schwann cells, and fibroblasts work together to create a working nerve. Schwann cells and neurons exchange molecular signals that regulate survival and differentiation. These signals are disrupted in CMT.[3]
Demyelinating Schwann cells causes abnormal axon structure and function. They may cause axon degeneration. Or they may simply cause axons to malfunction.[1]
The myelin sheath allows nerve cells to conduct signals faster. When the myelin sheath is damaged, nerve signals are slower, and this can be measured by a common neurological test, electromyography.
When the axon is damaged, on the other hand, this results in a reduced compound muscle action potential (CMAP).[4]
Symptoms
Symptoms of CMT usually begin in late childhood or early adulthood. Some people don't experience symptoms until their early thirties or forties. Usually, the initial symptom is foot drop early in the course of the disease. This can also cause claw toe, where the toes are always curled. Wasting of muscle tissue of the lower parts of the legs may give rise to "stork leg" or "inverted bottle" appearance. Weakness in the hands and forearms occurs in many people later in life as the disease progresses.
Symptoms and progression of the disease can vary. Breathing can be affected in some; so can hearing, vision, as well as the neck and shoulder muscles. Scoliosis is common. Hip sockets can be malformed. Gastrointestinal problems can be part of CMT, as can chewing, swallowing, and speaking (as vocal cords atrophy). A tremor can develop as muscles waste. Pregnancy has been known to exacerbate CMT, as well as extreme emotional stress. Patients with CMT must avoid periods of prolonged immobility such as when recovering from a secondary injury as prolonged periods of limited mobility can drastically accelerate symptoms of CMT.[5]
Neuropathic pain is often a symptom of CMT, though, like other symptoms of CMT, its presence and severity varies from case to case. For some people, pain can be significant to severe and interfere with daily life activities. However, pain is not experienced by all people with CMT. When pain is present as a symptom of CMT, it is comparable to that seen in other peripheral neuropathies, as well as Postherpetic neuralgia and Complex regional pain syndrome, among other diseases.[6]
Diagnosis
CMT can be diagnosed through symptoms, through measurement of the speed of nerve impulses (electromyography), through biopsy of the nerve, and through DNA testing. DNA testing can give a definitive diagnosis, but not all the genetic markers for CMT are known.CMT is first noticed when someone develops lower leg weakness and foot deformities such as foot drop, hammertoes and high arches. But signs alone do not lead to diagnosis. Patients must be referred to a neurologist or a physical medicine and rehabilitation physician (physiatrist). To see signs of muscle weakness the neurologist will ask patients to walk on their heels or to move part of their leg against an opposing force. In order to identify sensory loss the neurologist will test for deep tendon reflexes, such as the knee jerk, which are reduced or absent in CMT. The doctor will also ask about family history because CMT is hereditary. The lack of family history does not rule out CMT, but it will allow the doctor to rule out other causes of neuropathy such as diabetes or exposure to certain chemicals or drugs.[7]
In 2010, CMT was one of the first diseases where the genetic cause of a particular patient's disease was precisely determined by sequencing the whole genome of an affected individual.[8][9] Two mutations were identified in a gene, SH3TC2, known to cause CMT. Researchers then compared the affected patient's genome to the genomes of the patient's mother, father, and seven siblings with and without the disease. The mother and father each had one normal and one mutant copy of this gene, and had mild or no symptoms. The offspring that inherited two mutant genes presented fully with the disease. Sequencing the initial patient's whole genome cost $50,000, but researchers estimated that it would soon cost $5,000 and become common.
Types
As of early 2010, mutations in 39 genes have been identified as causes of CMT.[8] CMT can be categorized first into major clinical categories, and then into subtypes according to those mutations. Type 1 primarily affects the myelin sheath, and is either dominant, recessive or X-linked. Type 2 primarily affects the axon, and is either dominant or recessive. Other types are mixed.[9]
Clinical categories
Name Inheritance Frequency Notes CMT Type 1 (CMT1) Autosomal dominant Type 1 affects approximately one-third of CMT patients and is the most common type of CMT. The subtypes share clinical symptoms. Causes demyelination, which can be detected by measuring nerve conduction velocities. (anatomic changes directly affect the myelin sheath, with secondary axonal changes. In areas of focal demyelination, impulse conduction is slowed. The prolongation slows conduction velocity along the nerve segment.) CMT Type 2 (CMT2) Autosomal dominant
(except CMT2B1)Type 2 affects approximately 20-40% of CMT patients. Main effect is on the axon. The average nerve conduction velocity is slightly below normal, but generally above 38m/s The type-2 form of Charcot-Marie-Tooth disease (CMT2) tends to affect the lower extremities more than the upper extremities. CMT2 is often a clinically less severe disease than CMT1. CMT Type 3 (CMT3) Autosomal recessive Type 3 affects very few CMT patients. CMT Type 4 (CMT4) Autosomal recessive Type 4 affects very few CMT patients. CMT X-Linked (CMTX) X-linked dominant
(only CMTX1)CMTX affects approximately 10-20% of CMT patients. Approx 10% of X-linked CMT patients have some other form than CMTX. However a study published in 1997 indicates that a connexin 32 gene mutation is associated with this form which may be more common than previously thought.[10][11] Genetic subtypes
Type OMIM Gene Locus Description CMT1A 118220 PMP22 17p11.2 The most common form of the disease, 70-80% of Type 1 patients. Average NCV: 20-25m/s when associated with essential tremor and ataxia, called Roussy-Levy Syndrome 180800 CMT1B 118200 MPZ 1q22 Caused by mutations in the gene producing protein zero (P0). 5-10% of Type 1 patients. Average NCV: <15m/s CMT1C 601098 LITAF 16p13.1-p12.3 Causes severe demyelination, which can be detected by measuring nerve conduction velocities. Usually shows up in infancy. Average NCV: 26-42m/s. Identical symptoms to CMT-1A. CMT1D 607678 EGR2 10q21.1-q22.1 Average NCV: 15-20m/s CMT1E 118300 PMP22 17p11.2 Demyelinating, deafness CMT1F 607734 NEFL 8p21 CMT2A 118210 MFN2 or KIF1B 1p36 The cause is likely located on chromosome 1 for the mitofusion 2 protein. Some research has also linked this form of CMT to the protein kinesin 1B. Does not show up on nerve conduction velocity tests, because it is caused by axonopathy. CMT2B 600882 RAB7 (RAB7A, RAB7B) 3q21. CMT2B1 605588 LMNA 1q22 Axonal CMT (laminopathy) CMT2B2 605589 MED25 19q13.3 CMT2C 606071 'TRPV4 12q23-q24 May cause vocal cord, diaphragm, and distal weaknesses. CMT2D 601472 GARS 7p15 Patients with mutations in the GARS gene tend to have more severe symptoms in the upper extremities (hands), which is atypical for CMT in general. CMT2E 607684 NEFL 8p21 CMT2F 606595 HSPB1 7q11-q21 CMT2G 608591 12q12-13 CMT2H 607731 GDAP1 8q13-q21.1 CMT2J 607736 MPZ 1q22 CMT2K 607831 GDAP1 8q13-q21.1 CMT2L 608673 HSPB8 12q24 CMT3 145900 varies varies Sometimes called Dejerine-Sottas disease. Rarely found. Average NCV: Normal (50–60 m/s). This is an old classification. Currently this is referred to as CMT4F. CMT4A 214400 GDAP1 8q13-q21.1 CMT4B1 601382 MTMR2 11q22 CMT4B2 604563 CMT4B2 (SBF2) 11p15 May be called "SBF2/MTMR13". CMT4C 601596 KIAA1985 (SH3TC2) 5q32 May lead to respiratory compromise. CMT4D 601455 NDRG1 8q24.3 Demyelinating, deafness CMT4E 605253 EGR2 10q21.1-10q22.1 "CMT4E" is a tentative name CMT4F 145900 PRX 19q13.1-19q13.2 "CMT4F" is a tentative name CMT4H 609311 FGD4 12p11.21 CMT4J 611228 KIAA0274 (FIG4) 6q21 CMTX1 302800 GJB1 Xq13.1 Average NCV: 25-40m/s CMTX2 302801 Xq22.2 CMTX3 302802 Unknown, but 11 of 15 eliminated[12] Xq26 CMTX4 310490 Xq24-q26.1 Known as Cowchock syndrome CMTX5 311070 Xq22-q24 Known as Rosenberg-Chutorian syndrome. Signs include optic atrophy, polyneuropathy and deafness Management and treatment
Although there is no current standard treatment, the use of ascorbic acid has been proposed, and has shown some benefit in animal models.[13] A clinical trial to determine the effectiveness of high doses of ascorbic acid (vitamin C) in treating humans with CMT type 1A has been conducted.[14] The results of the trial upon children have shown that a high dosage intake of ascorbic acid is safe but the efficacy endpoints expected were not met.[15] In 2010, a study published in the Journal Science indicated that scientists had identified those proteins that control the thickness of myelin sheath. This discovery is expected to open the avenue to new treatments in the coming years.[16]
The most important activity for patients with CMT is to maintain what movement, muscle strength and flexibility they have. Therefore, physical therapy and moderate activity are recommended but overexertion should be avoided. A physiotherapist should be involved in designing a exercise program that fits a patient’s personal strengths and flexibility. Bracing can also be used to correct problems caused by CMT. Gait abnormalities can be corrected by the use of either articulated (hinged) or unarticulated, braces called AFOs (ankle-foot orthoses). These braces help control foot drop and ankle instability and often provide a better sense of balance for patients. Appropriate footwear is also very important for people with CMT, but they often have difficulty finding well-fitting shoes because of their high arched feet and hammer toes. Due to the lack of good sensory reception in the feet, CMT patients may also need to see a podiatrist for help in trimming nails or removing calluses that develop on the pads of the feet. A final decision a patient can make is to have surgery. Using a podiatrist or an orthopedic surgeon, patients can choose to stabilize their feet or correct progressive problems. These procedures include straightening and pinning the toes, lowering the arch, and sometimes, fusing the ankle joint to provide stability.[17]
The Charcot-Marie-Tooth Association classifies the chemotherapy drug vincristine as a "definite high risk" and states that "vincristine has been proven hazardous and should be avoided by all CMT patients, including those with no symptoms."[18]
There are also several corrective surgical procedures that can be done to improve physical condition.
Genetic testing is available for many of the different types of Charcot-Marie-Tooth and may help guide treatment.
History
The disease is named after those who classically described it: Jean-Martin Charcot (1825–1893), his pupil Pierre Marie (1853–1940) ("Sur une forme particulière d'atrophie musculaire progressive, souvent familiale débutant par les pieds et les jambes et atteignant plus tard les mains", Revue médicale, Paris, 1886; 6: 97-138.), and Howard Henry Tooth (1856–1925) ("The peroneal type of progressive muscular atrophy", dissertation, London, 1886.)
See also
References
- ^ a b c Krajewski, K. M. (2000). "Neurological dysfunction and axonal degeneration in Charcot-Marie-Tooth disease type 1A". Brain 123 (7): 1516–27. doi:10.1093/brain/123.7.1516.
- ^ Baloh, R. H.; Schmidt, R. E.; Pestronk, A.; Milbrandt, J. (2007). "Altered Axonal Mitochondrial Transport in the Pathogenesis of Charcot-Marie-Tooth Disease from Mitofusin 2 Mutations". Journal of Neuroscience 27 (2): 422–30. doi:10.1523/JNEUROSCI.4798-06.2007. PMID 17215403.
- ^ a b Berger, Philipp; Young, Peter; Suter, Ueli (2002). "Molecular cell biology of Charcot-Marie-Tooth disease". Neurogenetics 4 (1): 1–15. doi:10.1007/s10048-002-0130-z. PMID 12030326.
- ^ Yiu, Eppie M.; Burns, Joshua; Ryan, Monique M.; Ouvrier, Robert A. (2008). "Neurophysiologic abnormalities in children with Charcot-Marie-Tooth disease type 1A". Journal of the Peripheral Nervous System 13 (3): 236–241. doi:10.1111/j.1529-8027.2008.00182.x. PMID 18844790.
- ^ "Treatment and Management of CMT" (Press release). Charcot-Marie-Tooth Association. October 6, 2010. http://www.cmtausa.org/index.php?option=com_content&view=article&id=74:treatment-and-management-of-cmt&catid=10:resources&Itemid=51. Retrieved August 26, 2011.
- ^ Carter, Gregory T.; Jensen, Mark P.; Galer, Bradley S.; Kraft, George H.; Crabtree, Linda D.; Beardsley, Ruth M.; Abresch, Richard T.; Bird, Thomas D. (1998). "Neuropathic pain in Charcot-Marie-tooth disease". Archives of Physical Medicine and Rehabilitation 79 (12): 1560–4. doi:10.1016/S0003-9993(98)90421-X. PMID 9862301.
- ^ http://www.charcot-marie-tooth.org/about_cmt/diagnosis.php
- ^ a b Wade, Nicholas (2010-03-10). "Disease Cause Is Pinpointed With Genome". http://www.nytimes.com/2010/03/11/health/research/11gene.html.
- ^ a b Lupski, James R.; Reid, Jeffrey G.; Gonzaga-Jauregui, Claudia; Rio Deiros, David; Chen, David C.Y.; Nazareth, Lynne; Bainbridge, Matthew; Dinh, Huyen et al. (2010). "Whole-Genome Sequencing in a Patient with Charcot–Marie–Tooth Neuropathy". New England Journal of Medicine 362 (13): 1181–91. doi:10.1056/NEJMoa0908094. PMID 20220177.
- ^ Latour, Philippe; Fabreguette, Anne; Ressot, Catherine; Blanquet-Grossard, FranÇOise; Antoine, Jean-Christophe; Calvas, Patrick; Chapon, FranÇOise; Corbillon, Emmanuel et al. (1997). "New Mutations in the X-Linked Form of Charcot-Marie-Tooth Disease". European Neurology 37 (1): 38–42. doi:10.1159/000117403. PMID 9018031.
- ^ Abrams, Charles K.; Rash, John E. (2009). "Connexins in the Nervous System". In Harris, Andrew; Locke, Darren. Connexins. New York: Springer. pp. 323–57. doi:10.1007/978-1-59745-489-6_15. ISBN 978-1-934115-46-6. http://www.springerlink.com/content/978-1-59745-489-6#section=129548&page=1&locus=0.
- ^ Brewer M, Changi F, Antonellis A et al. (July 2008). "Evidence of a founder haplotype refines the X-linked Charcot-Marie-Tooth (CMTX3) locus to a 2.5 Mb region". Neurogenetics 9 (3): 191–5. doi:10.1007/s10048-008-0126-4. PMID 18458969.
- ^ Passage, Edith; Norreel, Jean Chrétien; Noack-Fraissignes, Pauline; Sanguedolce, Véronique; Pizant, Josette; Thirion, Xavier; Robaglia-Schlupp, Andrée; Pellissier, Jean François et al. (2004). "Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot-Marie-Tooth disease". Nature Medicine 10 (4): 396–401. doi:10.1038/nm1023. PMID 15034573.
- ^ "Clinical Trials - Neuromuscular Trial/Study". 2007-07-18. http://www.mda.org/research/view_ctrial.aspx?id=186. Retrieved 2008-05-28.
- ^ Burns, Joshua; Ouvrier, Robert A; Yiu, Eppie M; Joseph, Pathma D; Kornberg, Andrew J; Fahey, Michael C; Ryan, Monique M (2009). "Ascorbic acid for Charcot–Marie–Tooth disease type 1A in children: A randomised, double-blind, placebo-controlled, safety and efficacy trial". The Lancet Neurology 8 (6): 537–44. doi:10.1016/S1474-4422(09)70108-5.
- ^ "Nerves Under Control: Potential Treatment for Charcot-Marie-Tooth Disease". Science Daily. May 18, 2010. http://www.sciencedaily.com/releases/2010/05/100518112649.htm.
- ^ "Treatment and Management of CMT". http://www.cmtausa.org/index.php?option=com_content&view=article&id=74&catid=10&Itemid=51.
- ^ CMT Association: Medical Alert
External Links
- GeneReviews/NIH/NCBI/UW entry on Charcot-Marie-Tooth Hereditary Neuropathy Overview
- GeneReviews/NCBI/NIH/UW entry on Charcot-Marie-Tooth Neuropathy Type 1
- GeneReviews/NIH/NCBI/UW entry on Charcot-Marie-Tooth Neuropathy X Type 5
- OMIM entries on on Charcot-Marie-Tooth Neuropathy X Type 5
- GeneReviews/NCBI/NIH/UW entry on Charcot-Marie-Tooth Neuropathy X Type 1
- OMIM entries on Charcot-Marie-Tooth Neuropathy X Type 1
- GeneReviews/NCBI/NIH/UW entry on GARS-Associated Axonal Neuropathy, Charcot-Marie-Tooth Neuropathy Type 2D, Distal Spinal Muscular Atrophy V
- Charcot–Marie–Tooth disease at the Open Directory Project
- CMT United Kingdom www.cmt.org.uk
- CMT Central - An Insight into CMT - www.charcotmarietooth.webs.com
Nervous system pathology, PNS, somatic (G50–G64, 350–357) Nerve, nerve root, plexus V (Trigeminal neuralgia, Anesthesia dolorosa) · VII (Facial nerve paralysis, Bell's palsy, Melkersson–Rosenthal syndrome, Parry–Romberg syndrome, Central seven) · XI (Accessory nerve disorder)Lower limbGeneralPolyneuropathies/Polyradiculoneuropathy Charcot–Marie–Tooth disease · Dejerine–Sottas disease · Refsum's disease · Hereditary spastic paraplegiaOtherVesicle formation lysosome/melanosome: HPS1-HPS7 (Hermansky–Pudlak syndrome) · LYST (Chédiak–Higashi syndrome) ·
COPII: SEC23A (Cranio–lenticulo–sutural dysplasia)
APC: AP1S2 (X-Linked mental retardation 59) · AP3B1 (Hermansky–Pudlak syndrome 2) · AP4M1 (CPSQ3)Rab Cytoskeleton Vesicle fusion synaptic vesicle: SNAP29 (CEDNIK syndrome) · STX11 (Hemophagocytic lymphohistiocytosis 4)
caveolae: CAV1 (Congenital generalized lipodystrophy 3) · CAV3 (Limb-girdle muscular dystrophy 2B, Long QT syndrome 9)
vacuolar protein sorting: VPS33B (ARC syndrome) · VPS13B (Cohen syndrome)
DYSF (Distal muscular dystrophy)see also vesicular transport proteins
B structural (perx, skel, cili, mito, nucl, sclr) · DNA/RNA/protein synthesis (drep, trfc, tscr, tltn) · membrane (icha, slcr, atpa, abct, othr) · transduction (iter, csrc, itra), trfkArrestin Myelin Pelizaeus–Merzbacher disease · Dejerine-Sottas disease · Charcot–Marie–Tooth disease 1B, 2JPulmonary surfactant Cell adhesion molecule IgSF CAM: OFC7
Cadherin: DSG1 (Striate palmoplantar keratoderma 1) · DSG2 (Arrhythmogenic right ventricular dysplasia 10) · DSG4 (LAH1) · DSC2 (Arrhythmogenic right ventricular dysplasia 11)
Integrin: see cell surface receptor deficienciesTetraspanin Other KIND1 (Kindler syndrome) · HFE (HFE hereditary hemochromatosis) · DYSF (Distal muscular dystrophy, Limb-girdle muscular dystrophy 2B)see also other cell membrane proteins
B structural (perx, skel, cili, mito, nucl, sclr) · DNA/RNA/protein synthesis (drep, trfc, tscr, tltn) · membrane (icha, slcr, atpa, abct, othr) · transduction (iter, csrc, itra), trfkDeficiencies of intracellular signaling peptides and proteins GTP-binding protein regulators GTPase-activating proteinMarinesco–Sjögren syndrome · Aarskog–Scott syndrome · Juvenile primary lateral sclerosis · X-Linked mental retardation 1G protein cAMP/GNAS1: Pseudopseudohypoparathyroidism · Progressive osseous heteroplasia · Pseudohypoparathyroidism · Albright's hereditary osteodystrophy · McCune–Albright syndrome
CGL 2RAS: HRAS (Costello syndrome) · KRAS (Noonan syndrome 3, KRAS Cardiofaciocutaneous syndrome)
RAB: RAB7 (Charcot–Marie–Tooth disease) · RAB23 (Carpenter syndrome) · RAB27 (Griscelli syndrome type 2)
RHO: RAC2 (Neutrophil immunodeficiency syndrome)
ARF: SAR1B (Chylomicron retention disease) ARL13B (Joubert syndrome 8) · ARL6 (Bardet–Biedl syndrome 3)MAP kinase Other kinase/phosphatase RPS6KA3 (Coffin-Lowry syndrome) · CHEK2 (Li-Fraumeni syndrome 2) · IKBKG (Incontinentia pigmenti) · STK11 (Peutz–Jeghers syndrome) · DMPK (Myotonic dystrophy 1) · ATR (Seckel syndrome 1) · GRK1 (Oguchi disease 2) · WNK4/WNK1 (Pseudohypoaldosteronism 2)PTEN (Bannayan–Riley–Ruvalcaba syndrome, Lhermitte–Duclos disease, Cowden syndrome, Proteus-like syndrome) · MTM1 (X-linked myotubular myopathy) · PTPN11 (Noonan syndrome 1, LEOPARD syndrome, Metachondromatosis)Signal transducing adaptor proteins Other NF2 (Neurofibromatosis type II) · NOTCH3 (CADASIL) · PRKAR1A (Carney complex) · PRKAG2 (Wolff–Parkinson–White syndrome) · PRKCSH (PRKCSH Polycystic liver disease) · XIAP (XIAP2)see also intracellular signaling peptides and proteins
B structural (perx, skel, cili, mito, nucl, sclr) · DNA/RNA/protein synthesis (drep, trfc, tscr, tltn) · membrane (icha, slcr, atpa, abct, othr) · transduction (iter, csrc, itra), trfkCategories:- Neurological disorders
- Cytoskeletal defects
Wikimedia Foundation. 2010.