- Neurofibromatosis type II
-
Neurofibromatosis type II Classification and external resources ICD-10 D33, Q85.0 (ILDS Q85.020) ICD-9 237.72 OMIM 101000 DiseasesDB 8960 eMedicine neuro/496 radio/475 MeSH D016518  NF-2 Locus
NF-2 Locus
Neurofibromatosis Type II (or "MISME Syndrome", for "Multiple Inherited Schwannomas, Meningiomas, and Ependymomas") is an inherited disease. The main manifestation of the disease is the development of symmetric, non-malignant brain tumours in the region of the cranial nerve VIII, which is the auditory-vestibular nerve that transmits sensory information from the inner ear to the brain. Most people with this condition also experience problems in their eyes. NF II is caused by mutations of the "Merlin" gene,[1] which, it seems, influences the form and movement of cells. The principal treatments consist of neurosurgical removal of the tumors and surgical treatment of the eye lesions. There is no therapy for the underlying disorder of cell function caused by the genetic mutation.
Contents
Causes
Incidence, Mode of transmission, Epidemiology
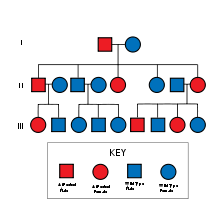 NF-2 may be inherited in an autosomal dominant fashion, as well as through random mutation.
NF-2 may be inherited in an autosomal dominant fashion, as well as through random mutation.NF II is an inheritable disorder with an autosomal dominant mode of transmission. Incidence of the disease is about 1 in 60,000.[2] There is a broad clinical spectrum known, but all patients checked have been found to have some mutation of the same gene on chromosome 22. Through statistics, it is suspected that one-half of cases are inherited, and one-half are the result of new, de novo mutations.
Pathogenesis, Molecular Biology and pathophysiological relations
NF II is caused by a defect in the gene that normally gives rise to a product called Merlin or Schwannomin, located on chromosome 22 band q11-13.1. This peptide is thought to have a tumor-suppressive function. In a normal cell, the concentrations of active (dephosphorylated) merlin are controlled by processes such as cell adhesion (which would indicate the need to restrain cell division). It is known that Merlin's deficiency can result in unmediated progression through the cell cycle due to the lack of contact-mediated tumour suppression, sufficient to result in the tumors characteristic of Neurofibromatosis type II. The NF II gene is presumed to result in either a failure to synthesize Merlin or the production of a defective peptide that lacks the normal tumor-suppressive effect. The Schwannomin-peptide consists of 595 amino acids. Comparison of Schwannomin with other proteins shows similarities to proteins that connect the cytoskeleton to the cell membrane. Mutations in the Schwannomin-gene are thought to alter the movement and shape of affected cells with loss of contact inhibition.
Pathology
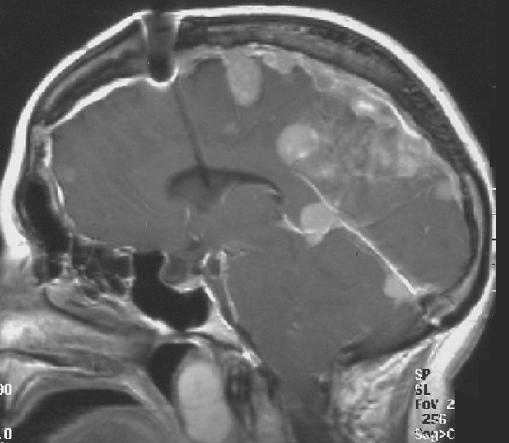
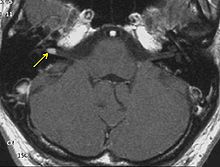 Schwannoma of the N. Vestibularis
Schwannoma of the N. Vestibularis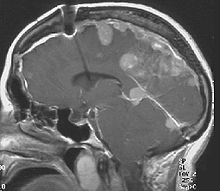 Meningeomas in a patient with NFII
Meningeomas in a patient with NFIIThe so-called acoustic neuroma of NF II is in fact a Schwannoma of the nervus vestibularis, or vestibular schwanomma. The misnomer of acoustic neuroma is still often used. The vestibular Schwannomas grow slowly at the inner entrance of the internal auditory meatus (meatus acousticus internus). They derive from the nerve sheaths of the upper part of the nervus vestibularis in the region between the central and peripheral myelin (Obersteiner-Redlich-Zone) within the area of the porus acousticus, 1 cm from the brainstem.
Genotype-Phenotype-Correlation
Many patients with NF II were included in studies that were designed to compare disease type and progression with exact determination of the associated mutation. The goal of such comparisons of genotype and phenotype is to determine whether specific mutations cause respective combinations of symptoms. This would be extremely valuable for the prediction of disease progression and the planning of therapy starting at a young age. The results of such studies are the following:
- In most cases the mutation in the NF II gene causes shortened peptides.
- There are no mutational hot-spots.
- Patients with Frameshift mutation- or Nonsense mutations suffer poor prognosis.
- Patients with Missense mutations have a better prognosis.
- In cases with Mutations in the splice-acceptor-region, there is no good correlation to determine.
- Point mutations may have only minor effects.
- Cases are published[citation needed] in which exactly the same mutation is associated with clearly different outcome.
These results suggest, that probably other factors (Environment, other mutations) will determine the clinical outcome.
Symptoms and Signs
The clinical spectrum of the disease is broad. In other words, people with NF II may develop a wide range of distinct problems.
- Acoustic nerve: 90% of the patients show bilateral acoustic neuromas on magnetic resonance imaging (MRI).
- Other cranial nerves and meninges: About 50% of patients develop tumours in other cranial nerves or Meningiomas.
- Spinal cord: About 50% of the patients develop spinal lesions. Only 40% of the spinal lesions are symptomatic. The spinal tumours in NF II are separated in two groups. Intramedullary lesions are located within the spinal tissue and usually belong to the so-called spinal astrocytomas or ependymomas. The extramedullary lesions are located within the small space between the surface of the spinal cord and the bony wall of the spinal canal. These tumours belong to the Schwannomas and Meningiomas.
- Skin: If children show neurofibromas, a diagnostic procedure should be performed to decide which form of neurofibromatosis causes the alterations.
- Eyes: Studies on patients with NF II show that more than 90% of the affected persons suffer eye lesions. The most common alteration in NF II is the juvenile subcapsular cataract (opacity of the lens) in young people.
"Presenting symptoms" (initial concern that brings a patient to a doctor) of a lesion of the nervus vestibulocochlearis due to a tumour in the region of the cerebello-pontine angle are the following: hearing loss (98%), tinnitus (70%), dysequilibrium (67%), headache (32%), facial numbness and weakness (29% and 10% respectively).
"Clinical signs" (alterations that are not regarded by the patient and that can be detected by the doctor in a clinical examination) of the lesion in discussion are: abnormal corneal reflex (33%), nystagmus (26%), facial hypesthesia (26%).
Evaluation (study of the patient with technical methods) shows the enlargement of the porus acousticus internus in the CT scan, enhancing tumours in the region of the cerebello-pontine angle in gadolinium-enhanced MRI scans, hearing loss in audiometric studies and perhaps pathological findings in Electronystagmography. Some times there are elevated levels of protein in liquor study.
Diagnosis
NF II can be diagnosed with 65% accuracy prenatally with chorionic villus sampling or amniocentesis.[3]
Bilateral acoustic neuromas are diagnostic of NF2.[4]
The diagnostic criteria for NF II are the following:
- Detection of bilateral acoustic neuroma by imaging-procedures
- First degree relative with NF II and the occurrence of Neurofibroma, Meningiomas, Glioma, or Schwannoma
- First degree relative with NF II and the occurrence of juvenile posterior subcapsular cataract.
The criteria have varied over time.[5]
Progression of the disease
In NF II Acoustic neuromas usually affect young people, whereas, in sporadic forms of Acoustic neuromas, the appearance of the tumour is limited to the elderly.
There are two forms of the NF II:[6]
- The Wishart-Phenotype is characterized by multiple cerebral and spinal lesions in patients younger than 20 years and with rapid progression of the tumours.
- Patients that develop single central tumours with slow progression after age of 20 are thought to have the Feiling-Gardner-Phenotype.
Treatment
Overview
Early diagnosis allows better planning of therapy in young patients with NF II. In many cases, the hearing loss is present for 10 years before the correct diagnosis is established. Early in the disease, surgery for an acoustic neurinoma can protect facial nerve function in many patients. In selected cases of patients with very small tumors and good bilateral hearing, surgery may offer the possibility of long-term hearing preservation.
Patients with the Wishard phenotype suffer multiple recurrences of the tumour after surgical treatment. In the case of facial nerve palsy, the muscles of the eyelids can lose their mobility, leading to conjunctivitis and corneal injury. "Lidloading" (implantation of small magnets, gold weights, or springs in the lid) can help prevent these complications. Other means of preserving corneal health include tarsorrhaphy, where the eyelids are partially sewn together to narrow the opening of the eye, or the use of punctal plugs, which block the duct that drains tears from the conjunctival sac. All these techniques conserve moisture from the lacrymal glands, which lubricates the cornea and prevents injury. Most patients with NF II develop cataracts, which often require replacement of the lens. Children of affected parents should have a specialist examination every year to detect developing tumors. Learning of sign-language is one means of preparation for those that will most probably suffer complete hearing loss.
The St. Louis Children's Hospital Neurofibromatosis Center maintains a comprehensive list of current NF research studies.
Operative therapy of acoustic neuroma
There are several different surgical techniques for the removal of acoustic neuroma.[7] The choice of approach is determined by size of the tumour, hearing capability, and general clinical condition of the patient.
- The retrosigmoid approach offers some opportunity for the retention of hearing.
- The translabyrinthine approach will sacrifice hearing on that side, but will usually spare the facial nerve. Post-Operative cerebrospinal fluid leaks are more common.
- The middle fossa approach is preferred for small tumours, and offers the highest probability of retention of hearing and vestibular function.
- Less invasive endoscopic techniques have been done outside of the United States for some time. Recovery times are reported to be faster. However, this technique is not yet mainstream among surgeons in the US.
Larger tumors can be treated by either the translabyrinthine approach or the retrosigmoid approach, depending upon the experience of the surgical team. With large tumors, the chance of hearing preservation is small with any approach. When hearing is already poor, the translabyrinthine approach may be used for even small tumors. Small, lateralized tumours in patients with good hearing should have the middle fossa approach. When the location of the tumour is more medial a retrosigmoid approach may be better.
Auditory canal decompression is another surgical technique that can prolong usable hearing when a vestibular schwannoma has grown too large to remove without damage to the cochlear nerve. In the IAC (internal auditory canal) decompression, a middle fossa approach is employed to expose the bony roof of the IAC without any attempt to remove the tumor. The bone overlying the acoustic nerve is removed, allowing the tumour to expand upward into the middle cranial fossa. In this way, pressure on the cochlear nerve is relieved, reducing the risk of further hearing loss from direct compression or obstruction of vascular supply to the nerve.
Radiosurgery is a conservative alternative to cranial base or other intracranial surgery. With conformal radiosurgical techniques, therapeutic radiation focused on the tumour, sparing exposure to surrounding normal tissues. Although radiosurgery can seldom completely destroy a tumor, it can often arrest its growth or reduce its size. While radiation is less immediately damaging than conventional surgery, it incurs a higher risk of subsequent malignant change in the irradiated tissues, and this risk in higher in NF2 than in sporadic (non-NF2) lesions.
Chemotherapy
A 2009 clinical trial at Massachusetts General Hospital used the cancer drug Bevacizumab (commercial name: Avastin) to treat 10 patients with neurofibromatosis type II. The result was published in The New England Journal of Medicine. Of the ten patients treated with bevacizumab, tumours shrank in 9 of them, with the median best response rate of 26%. Hearing improved in some of the patients, but improvements were not strongly correlated with tumour shrinkage. Bevacizumab works by cutting the blood supply to the tumours and thus depriving them of their growth vector. Side effects during the study included alanine aminotransferase, proteinuria, and hypertension (elevated blood pressure) among others.[8] A separate trial, published in The Neuro-oncology Journal, show 40% tumour reduction in the two patients with NF2, along with significant hearing improvement.[9]
Overall the researchers believed that bevacizumab showed clinically significant effects on NF2 patients. However, more research is needed before the full effects of bevacizumab can be established in NF2 patients.
Management of Hearing Loss in NF2
Because hearing loss in those with NF2 almost always occurs after acquisition of verbal language skills, patients do not always integrate well into the Deaf culture and are more likely to resort to auditory assistive technology. The most sophisticated of these devices is the cochlear implant, which can sometimes restore a high level of auditory function even when natural hearing is totally lost. However, the amount of destruction to the cochlear nerve caused by the typical NF2 schwannoma often precludes the use of such an implant. In these cases, an auditory brainstem implant (ABI) can restore a primitive level of hearing, which, when supplemented by lip reading, can restore a functional understanding of spoken language.
Sunitinib is being studied for treatment of meningioma which is associated with Neurofibromatosis.[10]
Drug therapy for vestibular schwannoma
Lapatinib is being studied by Jeffrey Allen at NYU Langone Medical Center for treatment of vestibular schwannoma in Neurofibromatsis type II.[11]
See also
References
- ^ Striedinger K, VandenBerg SR, Baia GS, McDermott MW, Gutmann DH, Lal A (November 2008). "The neurofibromatosis 2 tumor suppressor gene product, merlin, regulates human meningioma cell growth by signaling through YAP". Neoplasia 10 (11): 1204–12. PMC 2570596. PMID 18953429. http://www.neoplasia.com/abstract.php?msid=1779.
- ^ Evans DG (2009). "Neurofibromatosis type 2 (NF2): a clinical and molecular review". Orphanet J Rare Dis 4: 16. doi:10.1186/1750-1172-4-16. PMC 2708144. PMID 19545378. http://www.ojrd.com/content/4//16.
- ^ "Are there any prenatal tests for the neurofibromatoses?"
- ^ Christopher Gillberg (16 October 2003). Clinical Child Neuropsychiatry. Cambridge University Press. pp. 231–. ISBN 9780521543354. http://books.google.com/books?id=zxf4Z4PAiXwC&pg=PA231. Retrieved 20 December 2010.
- ^ "Neurofibromatosis Type 2: eMedicine Radiology". http://emedicine.medscape.com/article/342667-overview. Retrieved 2010-12-20.
- ^ Walter J, Kuhn SA, Brodhun M, Reichart R, Kalff R (June 2009). "Pulmonary meningioma and neurinoma associated with multiple CNS tumours in a patient with neurofibromatosis type 2". Clin Neurol Neurosurg 111 (5): 454–9. doi:10.1016/j.clineuro.2008.11.018. PMID 19249154. http://linkinghub.elsevier.com/retrieve/pii/S0303-8467(08)00407-1.
- ^ Jean Régis; Pierre-Hugues Roche (2008). Modern Management of Acoustic Neuroma. Karger Publishers. pp. 191–. ISBN 9783805583701. http://books.google.com/books?id=NJ_nYYYocKgC&pg=PA191. Retrieved 20 December 2010.
- ^ http://content.nejm.org/cgi/content/full/NEJMoa0902579v1
- ^ http://neuro-oncology.oxfordjournals.org/cgi/content/full/12/1/14
- ^ "Phase II Trial of Sunitinib (SU011248) in Patients With Recurrent or Inoperable Meningioma"
- ^ "Targeting auditory tumors", NYU Physician, Winter 2010-2011, page 5
External links
- General: http://www.neuroguide.com/
- GeneReview/NCBI/NIH/UW entry on Neurofibromatosis 2
- Society for Neuroscience
- Description of surgical management: Acoustic Neuroma Patient Archive
- Research/Advocacy: Advocure
- Information about auditory brainstem implants: House Ear Institute
- Research/Advocacy: Children's Tumor Foundation: Ending Neurofibromatosis Through Research Website
- NF KONTAKT.be (a nonprofit organization providing information and resources for families, Schools and Health Care workers dealing with NF1, NF2, and tumour-related neurofibromatosis in Belgium and providing awareness and support of Neurofibromatosis in Europe)
Phakomatosis (Q85, 759.5–759.6) Neurofibromatosis Angiomatosis Hamartoma Tuberous sclerosis · Hypothalamic hamartoma (Pallister-Hall syndrome) · Multiple hamartoma syndrome (Proteus syndrome, Cowden syndrome, Bannayan–Riley–Ruvalcaba syndrome, Lhermitte-Duclos disease)Other Abdallat Davis Farrage syndrome · Ataxia telangiectasia · Incontinentia pigmenti · Peutz–Jeghers syndromeNervous tissue tumors/NS neoplasm/Neuroectodermal tumor (ICD-O 9350–9589) (C70–C72, D32–D33, 191–192/225) Endocrine/
sellar (9350–9379)other: PinealomaCNS
(9380–9539)Astrocytoma (Pilocytic astrocytoma, Pleomorphic xanthoastrocytoma, Fibrillary (also diffuse or lowgrade) astrocytomas, Anaplastic astrocytoma, Glioblastoma multiforme)Multiple/unknownNeuroblastoma (Esthesioneuroblastoma, Ganglioneuroblastoma) · Medulloblastoma · Atypical teratoid rhabdoid tumorPrimitiveHematopoieticPNS: NST
(9540–9579)cranial and paraspinal nerves: Neurofibroma (Neurofibrosarcoma, Neurofibromatosis) · Neurilemmoma/Schwannoma (Acoustic neuroma) · Malignant peripheral nerve sheath tumornote: not all brain tumors are of nervous tissue, and not all nervous tissue tumors are in the brain (see brain metastases)
Categories:- Neurological disorders
- Neurosurgery
- Deficiencies of intracellular signaling peptides and proteins


Wikimedia Foundation. 2010.