- Pallister-Hall syndrome
-
Pallister-Hall syndrome Classification and external resources OMIM 146510 DiseasesDB 32013 MeSH D054975 Pallister-Hall syndrome is a disorder that affects the development of many parts of the body.
It is named for Judith Hall and Philip Pallister.[1][2]
Contents
Presentation
Most people with this condition have extra fingers and/or toes (polydactyly), and the skin between some fingers or toes may be fused (cutaneous syndactyly). An abnormal growth in the brain called a hypothalamic hamartoma is characteristic of this disorder. In many cases, these growths do not cause any medical problems; however, some hypothalamic hamartomas lead to seizures or hormone abnormalities that can be life-threatening in infancy. Other features of Pallister-Hall syndrome include a malformation of the airway called a bifid epiglottis, laryngeal cleft, an obstruction of the anal opening (imperforate anus), and kidney abnormalities. Although the signs and symptoms of this disorder vary from mild to severe, only a small percentage of affected people have serious complications.
Genetics
This condition is very rare; its prevalence is unknown. Mutations in the GLI3 gene cause Pallister-Hall syndrome. The GLI3 gene provides instructions for making a protein that controls gene expression, which is a process that regulates whether genes are turned on or off in particular cells. By interacting with certain genes at specific times during development, the GLI3 protein plays a role in the normal shaping (patterning) of many organs and tissues before birth. Defects in the same gene also cause Greig cephalopolysyndactyly syndrome.
Mutations that cause Pallister-Hall syndrome typically lead to the production of an abnormally short version of the GLI3 protein. Unlike the normal GLI3 protein, which can turn target genes on or off, the short protein can only turn off (repress) target genes. Researchers are working to determine how this change in the protein's function affects early development. It remains uncertain how GLI3 mutations can cause polydactyly, hypothalamic hamartoma, and the other features of Pallister-Hall syndrome.
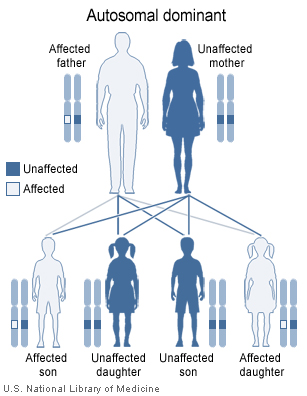
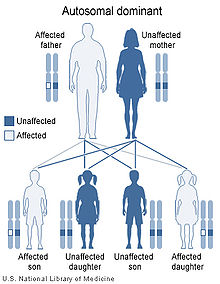 Pallister-Hall syndrome is inherited in an autosomal dominant pattern.
Pallister-Hall syndrome is inherited in an autosomal dominant pattern.
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. In some cases, an affected person inherits a mutation in the GLI3 gene from one affected parent. Other cases result from new mutations in the gene and occur in people with no history of the disorder in their family.
Seizures
As noted above, the hypothalamic hamartoma can cause seizures. The most common types of seizures that occur are known as Gelastic Epilepsy. The term Gelastic originates from the Greek word "Gelos" which means laughter. Seizures may begin at any age but usually before three or four years of age. The seizures usually start with laughter and the laughter is often described as being 'hollow' or 'empty' and not very pleasant. The laughter occurs suddenly, comes on for no obvious reason and is usually completely out of place. The most common areas of the brain which give rise to gelastic seizures are the hypothalamus (a small but extremely important structure deep in the centre of the brain), the temporal lobes and the frontal lobes. If the child has gelastic seizures and precocious puberty, then it is likely that the child will be found to have a hypothalamic hamartoma (a hamartoma in the hypothalamus part of the brain).
External links
{http://ghr.nlm.nih.gov/condition/pallister-hall-syndrome} {http://www.epilepsy.org.uk/info/gelastic.html}References
- ^ synd/506 at Who Named It?
- ^ Hall JG, Pallister PD, Clarren SK, et al (1980). "Congenital hypothalamic hamartoblastoma, hypopituitarism, imperforate anus and postaxial polydactyly--a new syndrome? Part I: clinical, causal, and pathogenetic considerations". Am. J. Med. Genet. 7 (1): 47–74. doi:10.1002/ajmg.1320070110. PMID 7211952.
- Pallister-Hall syndrome at NLM Genetics Home Reference
Genetic disorder, protein biosynthesis: Transcription factor/coregulator deficiencies (1) Basic domains 1.2: Feingold syndrome · Saethre-Chotzen syndrome
1.3: Tietz syndrome(2) Zinc finger
DNA-binding domains2.1 (Intracellular receptor): Thyroid hormone resistance · Androgen insensitivity syndrome (PAIS, MAIS, CAIS) · Kennedy's disease · PHA1AD pseudohypoaldosteronism · Estrogen insensitivity syndrome · X-linked adrenal hypoplasia congenita · MODY 1 · Familial partial lipodystrophy 3 · SF1 XY gonadal dysgenesis
2.2: Barakat syndrome · Tricho–rhino–phalangeal syndrome
2.3: Greig cephalopolysyndactyly syndrome/Pallister-Hall syndrome · Denys–Drash syndrome · Duane-radial ray syndrome · MODY 7 · MRX 89 · Townes–Brocks syndrome · Acrocallosal syndrome · Myotonic dystrophy 2
2.5: Autoimmune polyendocrine syndrome type 1(3) Helix-turn-helix domains 3.1: ARX (Ohtahara syndrome, Lissencephaly X2) · HLXB9 (Currarino syndrome) · HOXD13 (SPD1 Synpolydactyly) · IPF1 (MODY 4) · LMX1B (Nail–patella syndrome) · MSX1 (Tooth and nail syndrome, OFC5) · PITX2 (Axenfeld syndrome 1) · POU4F3 (DFNA15) · POU3F4 (DFNX2) · ZEB1 (Posterior polymorphous corneal dystrophy 3, Fuchs' dystrophy 3) · ZEB2 (Mowat-Wilson syndrome)
3.2: PAX2 (Papillorenal syndrome) · PAX3 (Waardenburg syndrome 1&3) · PAX4 (MODY 9) · PAX6 (Gillespie syndrome, Coloboma of optic nerve) · PAX8 (Congenital hypothyroidism 2) · PAX9 (STHAG3)
3.3: FOXC1 (Axenfeld syndrome 3, Iridogoniodysgenesis, dominant type) · FOXC2 (Lymphedema–distichiasis syndrome) · FOXE1 (Bamforth–Lazarus syndrome) · FOXE3 (Anterior segment mesenchymal dysgenesis) · FOXF1 (ACD/MPV) · FOXI1 (Enlarged vestibular aqueduct) · FOXL2 (Premature ovarian failure 3) · FOXP3 (IPEX)
3.5: IRF6 (Van der Woude syndrome, Popliteal pterygium syndrome)(4) β-Scaffold factors
with minor groove contacts4.2: Hyperimmunoglobulin E syndrome
4.3: Holt-Oram syndrome · Li-Fraumeni syndrome · Ulnar–mammary syndrome
4.7: Campomelic dysplasia · MODY 3 · MODY 5 · SF1 (SRY XY gonadal dysgenesis, Premature ovarian failure 7) · SOX10 (Waardenburg syndrome 4c, Yemenite deaf-blind hypopigmentation syndrome)
4.11: Cleidocranial dysostosis(0) Other transcription factors 0.6: Kabuki syndromeUngrouped Transcription coregulators Phakomatosis (Q85, 759.5–759.6) Neurofibromatosis Angiomatosis Hamartoma Tuberous sclerosis · Hypothalamic hamartoma (Pallister-Hall syndrome) · Multiple hamartoma syndrome (Proteus syndrome, Cowden syndrome, Bannayan–Riley–Ruvalcaba syndrome, Lhermitte-Duclos disease)Other Abdallat Davis Farrage syndrome · Ataxia telangiectasia · Incontinentia pigmenti · Peutz–Jeghers syndromeCategories:- Syndromes
- Autosomal dominant disorders
- Transcription factor deficiencies
Wikimedia Foundation. 2010.