- Greig cephalopolysyndactyly syndrome
-
Greig cephalopolysyndactyly syndrome Classification and external resources 
OMIM 175700 DiseasesDB 31558 Greig cephalopolysyndactyly syndrome is a disorder that affects development of the limbs, head, and face. The features of this syndrome are highly variable, ranging from very mild to severe. People with this condition typically have one or more extra fingers or toes (polydactyly) or an abnormally wide thumb or big toe (hallux).
The skin between the fingers and toes may be fused (cutaneous syndactyly). This disorder is also characterized by widely spaced eyes (ocular hypertelorism), an abnormally large head size (macrocephaly), and a high, prominent forehead. Rarely, affected individuals may have more serious medical problems including seizures, mental retardation, and developmental delay.
Contents
Pathophysiology
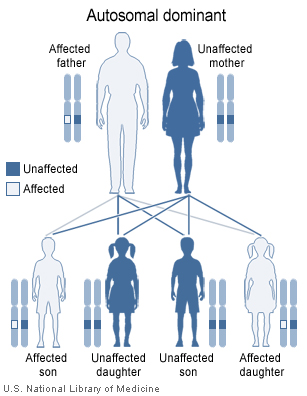
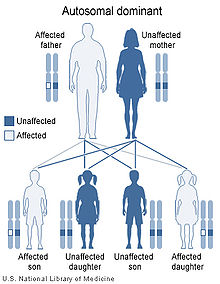 Greig cephalopolysyndactyly syndrome has an autosomal dominant pattern of inheritance.
Greig cephalopolysyndactyly syndrome has an autosomal dominant pattern of inheritance.
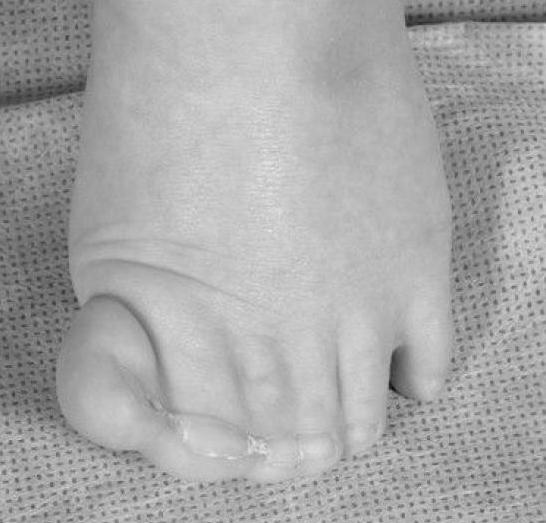
 The foot of the patient with Greig cephalopolysyndactyly shows a partially duplicated hallux with cutaneous syndactyly of several digits.
The foot of the patient with Greig cephalopolysyndactyly shows a partially duplicated hallux with cutaneous syndactyly of several digits. The hand of the patient with Greig cephalopolysyndactyly with syndactyly of several digits.
The hand of the patient with Greig cephalopolysyndactyly with syndactyly of several digits.Greig cephalopolysyndactyly syndrome is a chromosomal condition related to chromosome 7. Mutations in the GLI3 gene cause Greig cephalopolysyndactyly syndrome. The GLI3 gene provides instructions for making a protein that controls gene expression, which is a process that regulates whether genes are turned on or off in particular cells. By interacting with certain genes at specific times during development, the GLI3 protein plays a role in the normal shaping (patterning) of many organs and tissues before birth.
Different genetic changes involving the Gli3 gene can cause Greig cephalopolysyndactyly syndrome. In some cases, the condition results from a chromosomal abnormality, such as a large deletion or translocation of genetic material, in the region of chromosome 7 that contains the GLI3 gene. In other cases, a mutation in the GLI3 gene itself is responsible for the disorder. Each of these genetic changes prevents one copy of the gene in each cell from producing any functional protein. It remains unclear how a reduced amount of this protein disrupts early development and causes the characteristic features of Greig cephalopolysyndactyly syndrome.
This condition is inherited in an autosomal dominant pattern, which means the defective gene is located on an autosome, and only one copy of the defective GLI3 gene is sufficient to cause the disorder. In cases of dominant inheritance, an affected person inherits the genetic mutation or chromosomal abnormality from one affected parent.
Rare instances of this disorder are sporadic, and occur in people with no history of the condition in their family.
Eponym
The condition is named for David Middleton Greig.[1]
References
External links
- Greig cephalopolysyndactyly syndrome at NLM Genetics Home Reference
- GeneReview/NIH/UW entry on Greig Cephalopolysyndactyly Syndrome
Congenital malformations and deformations of musculoskeletal system / musculoskeletal abnormality (Q65–Q76, 754–756.3) Appendicular
limb / dysmeliahand deformity:Lowerhip:knee:Genu valgum · Genu varum · Genu recurvatum · Discoid meniscus · Congenital patellar dislocation · Congenital knee dislocationfoot deformity:Either / bothdactyly / digit:reduction deficits / limb:multiple joints:Axial Craniofacial dysostosis:other:Macrocephaly · Platybasia · Craniodiaphyseal dysplasia · Dolichocephaly · Greig cephalopolysyndactyly syndrome · Plagiocephaly · Saddle nosespinal curvature (Scoliosis) · Klippel-Feil syndrome · Spondylolisthesis · Spina bifida occulta · SacralizationThoracic skeletonribs:sternum:M: JNT
anat(h/c, u, t, l)/phys
noco(arth/defr/back/soft)/cong, sysi/epon, injr
proc, drug(M01C, M4)
Genetic disorder, protein biosynthesis: Transcription factor/coregulator deficiencies (1) Basic domains 1.2: Feingold syndrome · Saethre-Chotzen syndrome
1.3: Tietz syndrome(2) Zinc finger
DNA-binding domains2.1 (Intracellular receptor): Thyroid hormone resistance · Androgen insensitivity syndrome (PAIS, MAIS, CAIS) · Kennedy's disease · PHA1AD pseudohypoaldosteronism · Estrogen insensitivity syndrome · X-linked adrenal hypoplasia congenita · MODY 1 · Familial partial lipodystrophy 3 · SF1 XY gonadal dysgenesis
2.2: Barakat syndrome · Tricho–rhino–phalangeal syndrome
2.3: Greig cephalopolysyndactyly syndrome/Pallister-Hall syndrome · Denys–Drash syndrome · Duane-radial ray syndrome · MODY 7 · MRX 89 · Townes–Brocks syndrome · Acrocallosal syndrome · Myotonic dystrophy 2
2.5: Autoimmune polyendocrine syndrome type 1(3) Helix-turn-helix domains 3.1: ARX (Ohtahara syndrome, Lissencephaly X2) · HLXB9 (Currarino syndrome) · HOXD13 (SPD1 Synpolydactyly) · IPF1 (MODY 4) · LMX1B (Nail–patella syndrome) · MSX1 (Tooth and nail syndrome, OFC5) · PITX2 (Axenfeld syndrome 1) · POU4F3 (DFNA15) · POU3F4 (DFNX2) · ZEB1 (Posterior polymorphous corneal dystrophy 3, Fuchs' dystrophy 3) · ZEB2 (Mowat-Wilson syndrome)
3.2: PAX2 (Papillorenal syndrome) · PAX3 (Waardenburg syndrome 1&3) · PAX4 (MODY 9) · PAX6 (Gillespie syndrome, Coloboma of optic nerve) · PAX8 (Congenital hypothyroidism 2) · PAX9 (STHAG3)
3.3: FOXC1 (Axenfeld syndrome 3, Iridogoniodysgenesis, dominant type) · FOXC2 (Lymphedema–distichiasis syndrome) · FOXE1 (Bamforth–Lazarus syndrome) · FOXE3 (Anterior segment mesenchymal dysgenesis) · FOXF1 (ACD/MPV) · FOXI1 (Enlarged vestibular aqueduct) · FOXL2 (Premature ovarian failure 3) · FOXP3 (IPEX)
3.5: IRF6 (Van der Woude syndrome, Popliteal pterygium syndrome)(4) β-Scaffold factors
with minor groove contacts4.2: Hyperimmunoglobulin E syndrome
4.3: Holt-Oram syndrome · Li-Fraumeni syndrome · Ulnar–mammary syndrome
4.7: Campomelic dysplasia · MODY 3 · MODY 5 · SF1 (SRY XY gonadal dysgenesis, Premature ovarian failure 7) · SOX10 (Waardenburg syndrome 4c, Yemenite deaf-blind hypopigmentation syndrome)
4.11: Cleidocranial dysostosis(0) Other transcription factors 0.6: Kabuki syndromeUngrouped Transcription coregulators Categories:- Congenital disorders of musculoskeletal system
- Syndromes
- Transcription factor deficiencies
- Autosomal dominant disorders


Wikimedia Foundation. 2010.