- Crouzon syndrome
-
Crouzon syndrome Classification and external resources ICD-10 Q75.1 ICD-9 756.0 OMIM 123500 DiseasesDB 3203 eMedicine ped/511 derm/734 MeSH D003394 Crouzon syndrome is a genetic disorder known as a branchial arch syndrome. Specifically, this syndrome affects the first branchial (or pharyngeal) arch, which is the precursor of the maxilla and mandible. Since the branchial arches are important developmental features in a growing embryo, disturbances in their development create lasting and widespread effects.
Contents
Overview
This syndrome is named after Octave Crouzon,[1][2] a French physician who first described this disorder. He noted the affected patients were a mother and her daughter, implying a genetic basis. First called "craniofacial dysostosis", the disorder was characterized by a number of clinical features. This syndrome is caused by a mutation in the fibroblast growth factor receptor II, located on chromosome 10.
Breaking down the name, "craniofacial" refers to the skull and face, and "dysostosis" refers to malformation of bone.
Now known as Crouzon syndrome, the disease can be described by the rudimentary meanings of its former name. What occurs in the disease is that an infant's skull and facial bones, while in development, fuse early or are unable to expand. Thus, normal bone growth cannot occur. Fusion of different sutures leads to different patterns of growth of the skull. Examples include: trigonocephaly (fusion of the metopic suture), brachycephaly (fusion of the coronal suture), dolichocephaly (fusion of the sagittal suture), plagiocephaly (unilateral premature closure of lambdoid and coronal sutures), oxycephaly (fusion of coronal and lambdoidal sutures), Kleeblattschaedel (premature closure of all sutures).
Causes
Associations with mutations in the genes of FGFR2[3] and FGFR3[4] have been identified.[5] [6]
Heredity
Crouzon syndrome is autosomal dominant; children of a patient have a 50% chance of being affected.
Symptoms
As a result of the changes to the developing embryo, the symptoms are very pronounced features, especially in the face. Low-set ears are a typical characteristic, as in all of the disorders which are called branchial arch syndromes. The reason for this abnormality is that ears on a fetus are much lower than those on an adult. During normal development, the ears "travel" upward on the head; however, in Crouzon patients, this pattern of development is disrupted. Ear canal malformations are extremely common, generally resulting in some hearing loss. In particularly severe cases, Ménière's disease may occur.
The most notable characteristic of Crouzon syndrome is cranial synostosis, as described above, but it usually presents as brachycephaly, which results in the appearance of a short and broad head. Exophthalmos (bulging eyes due to shallow eye sockets after early fusion of surrounding bones), hypertelorism (greater than normal distance between the eyes), and psittichorhina (beak-like nose) are also symptoms. Additionally, a common occurrence is external strabismus, which can be thought of as opposite from the eye position found in Down syndrome. Lastly, hypoplastic maxilla (insufficient growth of the midface) results in relative mandibular prognathism (chin appears to protrude despite normal growth of mandible) and gives the effect of the patient having a concave face. Crouzon syndrome is also associated with PDA( patent ductus arteriosus) and aortic coarctation.
For reasons that are not entirely clear, most Crouzon patients also have noticeably shorter humerus and femur bones relative to the rest of their bodies than members of the general population. A small percentage of Crouzon patients also have what is called "Type II" Crouzon syndrome, distinguished by partial syndactyly.
Diagnosis
Diagnosis of Crouzon syndrome usually can occur at birth by assessing the signs and symptoms of the baby. Further analysis, including radiographs, magnetic resonance imaging (MRI) scans, genetic testing, X-rays and CT scans can be used to confirm the diagnosis of Crouzon syndrome.
Incidence
Incidence of Crouzon syndrome is currently estimated to occur in 1 out of every 25,000 people out of the general population.[citation needed] There is a greater frequency in families with a history of the disorder, but that doesn't mean that everyone in the family is affected (as referred to above).
Treatment
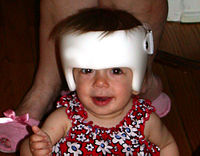 A child with Crouzon syndrome wearing a corrective cranial band.
A child with Crouzon syndrome wearing a corrective cranial band.
Surgery is typically used to prevent the closure of sutures of the skull from damaging the brain's development. Without surgery, blindness and mental retardation are typical outcomes. Craniofacial surgery is a discipline of plastic surgery. To move the orbits forward, craniofacial surgeons expose the skull and orbits and reshape the bone. To treat the midface deficiency, craniofacial surgeons can move the lower orbit and midface bones forward For jaw surgery, either plastic surgeons or oral and maxillofacial (OMS) surgeons have experience to perform these operations. It is rare to wear a custom-fitted helmet (or cranial band) for several months after surgery as that is only for single-suture "strip craniectomy" repair. Crouzon patients tend to have multiple sutures involved, most specifically bilateral coronal craniosynostoses, therefore an open operation is used rather than the strip craniectomy with helmeting.
Once treated for the cranial vault symptoms, Crouzon patients generally go on to live a normal lifespan.
Dental significance
For dentists, this disorder is important to understand since many of the physical abnormalities are present in the head, and particularly the oral cavity. Common features are a narrow/high-arched palate, posterior bilateral crossbite, hypodontia (missing some teeth), and crowding of teeth. Due to maxillary hypoplasia, Crouzon patients generally have a considerable permanent underbite and subsequently cannot chew using their incisors. For this reason, Crouzon patients sometimes eat in an unusual way--eating fried chicken with a fork, for example, or breaking off pieces of a sandwich rather than taking a bite in it.
See also
References
- ^ synd/1383 at Who Named It?
- ^ L. E. O. Crouzon. Dysostose cranio-faciale héréditaire. Bulletin de la Société des Médecins des Hôpitaux de Paris, 1912, 33: 545-555.
- ^ Reardon W, Winter RM, Rutland P, Pulleyn LJ, Jones BM, Malcolm S (September 1994). "Mutations in the fibroblast growth factor receptor 2 gene cause Crouzon syndrome". Nat. Genet. 8 (1): 98–103. doi:10.1038/ng0994-98. PMID 7987400.
- ^ Meyers GA, Orlow SJ, Munro IR, Przylepa KA, Jabs EW (December 1995). "Fibroblast growth factor receptor 3 (FGFR3) transmembrane mutation in Crouzon syndrome with acanthosis nigricans". Nat. Genet. 11 (4): 462–4. doi:10.1038/ng1295-462. PMID 7493034.
- ^ James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology. (10th ed.). Saunders. ISBN 0-7216-2921-0.
- ^ Vajo Z, Francomano CA, Wilkin DJ. (February 2000). "The molecular and genetic basis of fibroblast growth factor receptor 3 disorders: the achondroplasia family of skeletal dysplasias, Muenke craniosynostosis, and Crouzon syndrome with acanthosis nigricans". Endocrine Reviews 21 (1): 23–39. doi:10.1210/er.21.1.23. PMID 10696568.
External links
- GeneReviews/NIH/NCBI/UW entry on FGFR-Related Craniosynostosis Syndromes
- Crouzon Syndrome - About.com
- Crouzon Syndrome - Seattle Children's Hospital Craniofacial Center
- cleftAdvocate - Non-profit support organization for all craniofacial conditions; on-line and in-person family support, insurance and advocacy assistance, and more.
- General Information at hopkinsmedicine.org
Congenital malformations and deformations of musculoskeletal system / musculoskeletal abnormality (Q65–Q76, 754–756.3) Appendicular
limb / dysmeliahand deformity:Lowerhip:knee:Genu valgum · Genu varum · Genu recurvatum · Discoid meniscus · Congenital patellar dislocation · Congenital knee dislocationfoot deformity:Either / bothdactyly / digit:reduction deficits / limb:multiple joints:Axial Craniofacial dysostosis:other:spinal curvature (Scoliosis) · Klippel-Feil syndrome · Spondylolisthesis · Spina bifida occulta · SacralizationThoracic skeletonribs:sternum:M: JNT
anat(h/c, u, t, l)/phys
noco(arth/defr/back/soft)/cong, sysi/epon, injr
proc, drug(M01C, M4)
Categories:- Dental disorders
- Genodermatoses
- Syndromes
- Congenital disorders of musculoskeletal system
- Cell surface receptor deficiencies
Wikimedia Foundation. 2010.