- Familial hypercholesterolemia
-
Familial hypercholesterolemia Classification and external resources 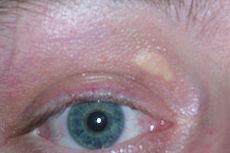
Xanthelasma palpebrarum, yellowish patches consisting of cholesterol deposits above the eyelids. These are more common in people with FH.ICD-10 E78.0 ICD-9 272.0 OMIM 143890 DiseasesDB 4707 MedlinePlus 000392 eMedicine med/1072 MeSH D006938 Familial hypercholesterolemia (abbreviated FH, also spelled familial hypercholesterolaemia) is a genetic disorder characterized by high cholesterol levels, specifically very high levels of low-density lipoprotein (LDL, "bad cholesterol"), in the blood and early cardiovascular disease. Many patients have mutations in the LDLR gene that encodes the LDL receptor protein, which normally removes LDL from the circulation, or apolipoprotein B (ApoB), which is the part of LDL that binds with the receptor; mutations in other genes are rare. Patients who have one abnormal copy (are heterozygous) of the LDLR gene may have premature cardiovascular disease at the age of 30 to 40. Having two abnormal copies (being homozygous) may cause severe cardiovascular disease in childhood. Heterozygous FH is a common genetic disorder, occurring in 1:500 people in most countries; homozygous FH is much rarer, occurring in 1 in a million births.[1]
Heterozygous FH is normally treated with statins, bile acid sequestrants or other hypolipidemic agents that lower cholesterol levels. New cases are generally offered genetic counseling. Homozygous FH often does not respond to medical therapy and may require other treatments, including LDL apheresis (removal of LDL in a method similar to dialysis) and occasionally liver transplantation.[1]
Contents
Signs and symptoms
Physical signs
High cholesterol levels normally do not cause any symptoms. Cholesterol may be deposited in various places in the body that are visible from the outside, such as in yellowish patches around the eyelids (xanthelasma palpebrarum), the outer margin of the iris (arcus senilis corneae) and in the form of lumps in the tendons of the hands, elbows, knees and feet, particularly the Achilles tendon (tendon xanthoma).[1][2]
Cardiovascular disease
Accelerated deposition of cholesterol in the walls of arteries leads to atherosclerosis, the underlying cause of cardiovascular disease. The most common problem in FH is the development of coronary artery disease (atherosclerosis of the coronary arteries that supply the heart) at a much younger age than would be expected in the general population. This may lead to angina pectoris (chest pain or tightness on exertion) or heart attacks. Less commonly, arteries of the brain are affected; this may lead to transient ischemic attacks (brief episodes of weakness on one side of the body or inability to talk) or occasionally stroke. Peripheral artery occlusive disease (obstruction of the arteries of the legs) occurs mainly in people with FH who smoke; this can cause pain in the calf muscles during walking that resolves with rest (intermittent claudication) and problems due to a decreased blood supply to the feet (such as gangrene).[3]
Atherosclerosis risk is increased further with age and in those who smoke, have diabetes, high blood pressure and a family history of cardiovascular disease.[1][4]
Diagnosis
Lipid measurements
Cholesterol levels may be determined as part of health screening for health insurance or occupational health, when the external physical signs such as xanthelasma, xanthoma, arcus are noticed, symptoms of cardiovascular disease develop, or a family member has been found to have FH. A pattern compatible with hyperlipoproteinemia type IIa on the Fredrickson classification is typically found: raised level of total cholesterol, markedly raised level of low-density lipoprotein (LDL), normal level of high-density lipoprotein (HDL), and normal level of triglycerides. The LDL is typically above the 75th percentile, that is, 75% of the healthy population would have a lower LDL level.[1] Cholesterol levels can be drastically higher in FH patients who are also obese.[3]
Mutation analysis
On the basis of the isolated high LDL and clinical criteria (which differ by country), genetic testing for LDL receptor mutations and ApoB mutations can be performed. Mutations are detected in between 50 and 80% of cases; those without a mutation often have higher triglyceride levels and may in fact have other causes for their high cholesterol, such as combined hyperlipidemia due to metabolic syndrome.[5]
Differential diagnosis
FH needs to be distinguished from familial combined hyperlipidemia and polygenic hypercholesterolemia. Lipid levels and the presence of xanthomata can confirm the diagnosis. Sitosterolemia and cerebrotendineous xanthomatosis are two rare conditions that can also present with premature atherosclerosis and xanthomas. The latter condition can also involve neurological or psychiatric manifestations, cataracts, diarrhea and skeletal abnormalities.[6]
Genetics
The most common genetic defects in FH are LDLR mutations (prevalence 1 in 500, depending on the population), ApoB mutations (prevalence 1 in 1000), PCSK9 mutations (less than 1 in 2500) and LDLRAP1. The related disease sitosterolemia, which has many similarities with FH and also features cholesterol accumulation in tissues, is due to ABCG5 and ABCG8 mutations.[1]
LDL receptor
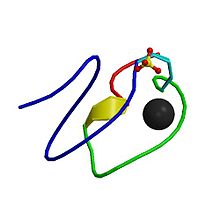 Schematic representation of the LDL receptor protein.
Schematic representation of the LDL receptor protein.
The LDL receptor gene is located on the short arm of chromosome 19 (19p13.1-13.3). It comprises 18 exons and spans 45 kb, and the protein gene product contains 839 amino acids in mature form. A single abnormal copy (heterozygote) of FH causes cardiovascular disease by the age of 50 in about 40% of cases. Having two abnormal copies (homozygote) causes accelerated atherosclerosis in childhood, including its complications. The plasma LDL levels are inversely related to the activity of LDL receptor (LDLR). Homozygotes have LDLR activity of less than 2%, while heterozygotes have defective LDL processing with receptor activity being 2–25%, depending on the nature of the mutation. Over 1000 different mutations are known.[1]
There are five major classes of FH due to LDLR mutations:[7]
- Class I: LDLR is not synthesized at all.
- Class II: LDLR is not properly transported from the endoplasmic reticulum to the Golgi apparatus for expression on the cell surface.
- Class III: LDLR does not properly bind LDL on the cell surface because of a defect in either apolipoprotein B100 (R3500Q) or in LDL-R.
- Class IV: LDLR bound to LDL does not properly cluster in clathrin-coated pits for receptor-mediated endocytosis.
- Class V: LDLR is not recycled back to the cell surface.
ApoB
ApoB, in its ApoB100 form, is the main apoprotein, or protein part of the lipoprotein particle. Its gene is located on the second chromosome (2p24-p23) and is between 21.08 and 21.12 Mb long. FH is often associated with the mutation of R3500Q, which causes replacement of arginine by glutamine at position 3500. The mutation is located on a part of the protein that normally binds with the LDL receptor, and binding is reduced as a result of the mutation. Like LDLR, the number of abnormal copies determines the severity of the hypercholesterolemia.[1][8]
PCSK9
Mutations in the proprotein convertase subtilisin/kexin type 9 (PCSK9) gene were linked to autosomal dominant (i.e. requiring only one abnormal copy) FH in a 2003 report.[1][9] The gene is located on the first chromosome (1p34.1-p32) and encodes a 666 amino acid protein that is expressed in the liver. It has been suggested that PCSK9 causes FH mainly by reducing the number of LDL receptors on liver cells.[10]
LDLRAP1
Abnormalities in the ARH gene, also known as LDLRAP1, were first reported in a family in 1973.[11] In contrast to the other causes, two abnormal copies of the gene are required for FH to develop (autosomal recessive). The mutations in the protein tend to cause the production of a shortened protein. Its real function is unclear, but it seems to play a role in the relation between the LDL receptor and clathrin-coated pits. Patients with autosomal recessive hypercholesterolemia tend to have more severe disease than LDLR-heterozygotes but less severe than LDLR-homozygotes.[1]
Pathophysiology
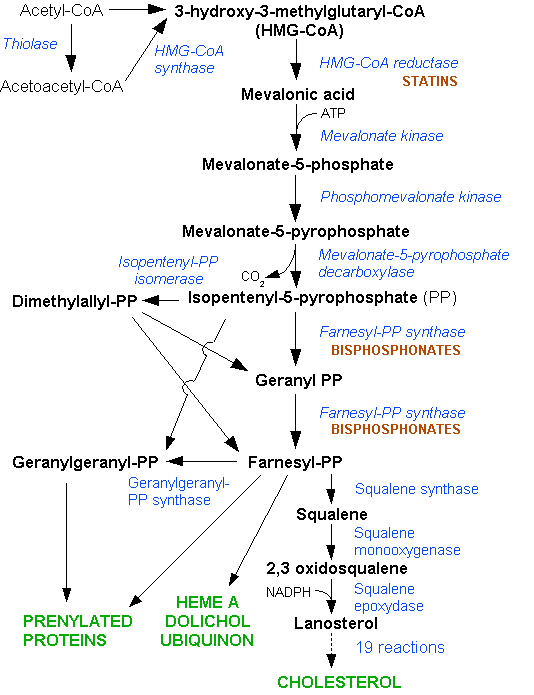
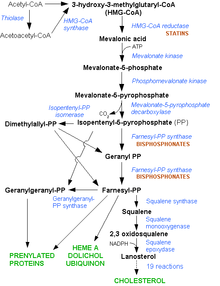 Cholesterol is synthesized in the HMG-CoA reductase pathway.Main articles: LDL receptor and atherosclerosis
Cholesterol is synthesized in the HMG-CoA reductase pathway.Main articles: LDL receptor and atherosclerosisLDL cholesterol normally circulates in the body for 2.5 days, and subsequently binds to the LDL receptor on the liver cells, undergoes endocytosis, and is digested. LDL is removed, and synthesis of cholesterol by the liver is suppressed in the HMG-CoA reductase pathway.[12] In FH, LDL receptor function is reduced or absent, and LDL circulates for an average duration of 4.5 days, resulting in significantly increased level of LDL cholesterol in the blood with normal levels of other lipoproteins.[3] In mutations of ApoB, reduced binding of LDL particles to the receptor causes the increased level of LDL cholesterol. It is not known how the mutation causes LDL receptor dysfunction in mutations of PCSK9 and ARH.[1]
Although atherosclerosis occurs to a certain degree in all people, FH patients may develop accelerated atherosclerosis due to the excess level of LDL. The degree of atherosclerosis approximately depends of the number of LDL receptors still expressed and the functionality of these receptors. In many heterozygous forms of FH, the receptor function is only mildly impaired, and LDL levels will remain relatively low. In the more serious homozygous forms, the receptor is not expressed at all.[1]
Some studies of FH cohorts suggest that additional risk factors are generally at play when an FH patient develops atherosclerosis.[13][14] In addition to the classic risk factors such as smoking, high blood pressure, and diabetes, genetic studies have shown that a common abnormality in the prothrombin gene (G20210A) increases the risk of cardiovascular events in patients with FH.[15] Several studies found that a high level of lipoprotein(a) was an additional risk factor for ischemic heart disease.[16][17] The risk was also found to be higher in patients with a specific genotype of the angiotensin-converting enzyme (ACE).[18]
Screening
Although case finding among family members of patients with known FH is a cost-effective approach, other strategies such as universal screening at the age of 16 have also been suggested.[19][20] The latter approach may however be less cost-effective in the short term.[21] Screening at an age lower than 16 would lead to an unacceptably high rate of false positives.[3]
Treatment
Heterozygous FH
FH is usually treated with statins. Statins act by inhibiting the enzyme hydroxymethylglutaryl CoA reductase (HMG-CoA-reductase) in the liver. In response, the liver produces more LDL receptors, which remove circulating LDL from the blood. Statins effectively lower cholesterol and LDL levels, although sometimes add-on therapy with other drugs is required, such as bile acid sequestrants (cholestyramine or colestipol), nicotinic acid preparations or fibrates.[1] Control of other risk factors for cardiovascular disease is required, as risk remains somewhat elevated even when cholesterol levels are controlled. Professional guidelines recommend that the decision to treat an FH patient with statins should not be based on the usual risk prediction tools (such as those derived from the Framingham Heart Study), as they are likely to underestimate the risk of cardiovascular disease; unlike the rest of the population, FH have had high levels of cholesterol since birth, probably increasing their relative risk.[22] Prior to the introduction of the statins, clofibrate (an older fibrate that often caused gallstones), probucol (especially in large xanthomas) and thyroxine were used to reduce LDL cholesterol levels.
More controversial is the addition of ezetimibe, which inhibits cholesterol absorption in the gut. While it reduces LDL cholesterol, it does not appear to improve a marker of atherosclerosis called the intima-media thickness. Whether this means that ezetimibe is of no overall benefit in FH is unknown.[23]
There are no interventional studies that directly show mortality benefit of cholesterol lowering in FH patients. Rather, evidence of benefit is derived from a number of trials conducted in people who have polygenic hypercholesterolemia (in which heredity plays a smaller role). Still, an observational study of a large British registry showed that mortality in FH patients had started to improve in the early 1990s, when statins were introduced.[24]
Homozygous FH
Homozygous FH is harder to treat. The LDL receptors are minimally functional, if at all. Only high doses of statins, often in combination with other medications, are modestly effective in improving lipid levels.[25] If medical therapy is not successful at reducing cholesterol levels, LDL apheresis may be used; this filters LDL from the bloodstream in a process reminiscent of dialysis.[1] Very severe cases may be considered for a liver transplant; this provides a liver with normally functional LDL receptors, and leads to rapid improvement of the cholesterol levels, but at the risk of complications from any solid organ transplant (such as rejection, infections, or side-effects of the medication required to suppress rejection).[26][27] Other surgical techniques include partial ileal bypass surgery, in which part of the small bowel is bypassed to decrease the absorption of nutrients and hence cholesterol, and portacaval shunt surgery, in which the portal vein is connected to the vena cava to allowing blood with nutrients from the intestine to bypass the liver.[28][29][30]
Inhibition of the microsomal triglyceride transfer protein, for example with the investigational drug lomitapide, and infusion of recombinant human apolipoprotein A1 are being explored as medical treatment options.[31][32] Gene therapy is a possible future alternative.[33] Mipomersen is in phase 3 trials.
In children
Given that FH is present from birth and atherosclerotic changes may begin early in life,[34] it is sometimes necessary to treat adolescents or even teenagers with agents that were originally developed for adults. Due to safety concerns, many doctors prefer to use bile acid sequestrants and fenofibrate as these are licensed in children.[35] Nevertheless, statins seem safe and effective,[36][37] and in older children may be used as in adults.[3][35]
A multidisciplinary expert panel in 2006 advised on early combination therapy with LDL apheresis, statins and cholesterol absorption inhibitors in children with homozygous FH at the highest risk.[38]
Epidemiology
In most populations studied, heterozygous FH occurs in about 1:500 people, but not all develop symptoms.[1] Homozygous FH occurs in about 1:1,000,000.[1][3]
LDLR mutations are more common in certain populations, presumably because of a genetic phenomenon known as the founder effect—they were founded by a small group of individuals, one or several of whom was a carrier of the mutation. The Afrikaner, French Canadians, Lebanese Christians, and Finns have high rates of specific mutations that make FH particularly common in these groups. APOB mutations are more common in Central Europe.[1]
History
The Norwegian physician Dr C. Müller first associated the physical signs, high cholesterol levels and autosomal dominant inheritance in 1938.[39] In the early 1970s and 1980s, the genetic cause for FH was described by Dr Joseph L. Goldstein and Dr Michael S. Brown of Dallas, Texas. Initially, they found increased activity of HMG-CoA reductase, but studies showed that this did not explain the very abnormal cholesterol levels in FH patients.[40] The focus shifted to the binding of LDL to its receptor, and effects of impaired binding on metabolism; this proved to be the underlying mechanism for FH.[41] Subsequently numerous mutations in the protein were directly identified by sequencing.[7] They later won the 1985 Nobel Prize in Medicine for their discovery of the LDL receptor and its impact on lipoprotein metabolism.[42]
See also
- Primary hyperlipoproteinemia
- Familial hypertriglyceridemia
- Lipoprotein lipase deficiency
- Familial apoprotein CII deficiency
References
- ^ a b c d e f g h i j k l m n o p q Rader DJ, Cohen J, Hobbs HH (2003). "Monogenic hypercholesterolemia: new insights in pathogenesis and treatment". J. Clin. Invest. 111 (12): 1795–803. doi:10.1172/JCI18925. PMC 161432. PMID 12813012. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=161432. Full text at PMC: 161432
- ^ Tsouli SG, Kiortsis DN, Argyropoulou MI, Mikhailidis DP, Elisaf MS (2005). "Pathogenesis, detection and treatment of Achilles tendon xanthomas". Eur. J. Clin. Invest. 35 (4): 236–44. doi:10.1111/j.1365-2362.2005.01484.x. PMID 15816992.
- ^ a b c d e f Durrington P (2003). "Dyslipidaemia". Lancet 362 (9385): 717–31. doi:10.1016/S0140-6736(03)14234-1. PMID 12957096.
- ^ Jansen AC, van Aalst-Cohen ES, Tanck MW, et al. (2004). "The contribution of classical risk factors to cardiovascular disease in familial hypercholesterolaemia: data in 2400 patients". J. Intern. Med. 256 (6): 482–90. doi:10.1111/j.1365-2796.2004.01405.x. PMID 15554949.
- ^ van Aalst-Cohen ES, Jansen AC, Tanck MW, et al. (2006). "Diagnosing familial hypercholesterolaemia: the relevance of genetic testing". Eur. Heart J. 27 (18): 2240–6. doi:10.1093/eurheartj/ehl113. PMID 16825289. http://eurheartj.oxfordjournals.org/cgi/content/full/27/18/2240.
- ^ Moghadasian MH, Salen G, Frohlich JJ, Scudamore CH (April 2002). "Cerebrotendinous xanthomatosis: a rare disease with diverse manifestations". Arch. Neurol. 59 (4): 527–9. doi:10.1001/archneur.59.4.527. PMID 11939886. http://archneur.ama-assn.org/cgi/content/full/59/4/527.
- ^ a b Hobbs HH, Brown MS, Goldstein JL (1992). "Molecular genetics of the LDLR gene in familial hypercholesterolemia". Hum. Mutat. 1 (6): 445–66. doi:10.1002/humu.1380010602. PMID 1301956.
- ^ Vega GL, Grundy SM (1986). "In vivo evidence for reduced binding of low density lipoproteins to receptors as a cause of primary moderate hypercholesterolemia". J. Clin. Invest. 78 (5): 1410–4. doi:10.1172/JCI112729. PMC 423848. PMID 3771801. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=423848. Full text at PMC: 423848
- ^ Abifadel M, Varret M, Rabès JP, et al. (2003). "Mutations in PCSK9 cause autosomal dominant hypercholesterolemia". Nat. Genet. 34 (2): 154–6. doi:10.1038/ng1161. PMID 12730697.
- ^ Seidah NG, Khatib AM, Prat A (2006). "The proprotein convertases and their implication in sterol and/or lipid metabolism" (PDF). Biol. Chem. 387 (7): 871–7. doi:10.1515/BC.2006.110. PMID 16913836. http://www.atypon-link.com/WDG/doi/pdf/10.1515/BC.2006.110.
- ^ Khachadurian AK, Uthman SM (1973). "Experiences with the homozygous cases of familial hypercholesterolemia. A report of 52 patients". Nutr Metab 15 (1): 132–40. doi:10.1159/000175431. PMID 4351242.
- ^ Brown MS, Goldstein JL (1974). "Familial hypercholesterolemia: defective binding of lipoproteins to cultured fibroblasts associated with impaired regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity". Proc. Natl. Acad. Sci. U.S.A. 71 (3): 788–92. doi:10.1073/pnas.71.3.788. PMC 388099. PMID 4362634. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=388099. Full text at PMC: 388099
- ^ Scientific Steering Committee on behalf of the Simon Broome Register Group (1991). "Risk of fatal coronary heart disease in familial hypercholesterolaemia". BMJ 303 (6807): 893–6. doi:10.1136/bmj.303.6807.893. PMC 1671226. PMID 1933004. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1671226. Full text at PMC: 1671226
- ^ Sijbrands EJ, Westendorp RG, Defesche JC, de Meier PH, Smelt AH, Kastelein JJ (2001). "Mortality over two centuries in large pedigree with familial hypercholesterolaemia: family tree mortality study". BMJ 322 (7293): 1019–23. doi:10.1136/bmj.322.7293.1019. PMC 31037. PMID 11325764. http://www.bmj.com/cgi/content/full/322/7293/1019. Full text at PMC: 31037
- ^ Jansen AC, van Aalst-Cohen ES, Tanck MW, et al. (2005). "Genetic determinants of cardiovascular disease risk in familial hypercholesterolemia". Arterioscler. Thromb. Vasc. Biol. 25 (7): 1475–81. doi:10.1161/01.ATV.0000168909.44877.a7. PMID 15879303. http://atvb.ahajournals.org/cgi/content/full/25/7/1475.
- ^ Wiklund O, Angelin B, Olofsson SO, Eriksson M, Fager G, Berglund L, Bondjers G (Jun 1990). "Apolipoprotein(a) and ischaemic heart disease in familial hypercholesterolaemia" (Free full text). Lancet 335 (8702): 1360–1363. doi:10.1016/0140-6736(90)91242-3. PMID 1971660. http://toxnet.nlm.nih.gov/cgi-bin/sis/search/r?dbs+hsdb:@term+@rn+111-14-8.
- ^ Seed M, Hoppichler F, Reaveley D, Mccarthy S, Thompson GR, Boerwinkle E, Utermann G (May 1990). "Relation of serum lipoprotein(a) concentration and apolipoprotein(a) phenotype to coronary heart disease in patients with familial hypercholesterolemia" (Free full text). The New England journal of medicine 322 (21): 1494–1499. doi:10.1056/NEJM199005243222104. ISSN 0028-4793. PMID 2139920. http://ClinicalTrials.gov/search/term=2139920%20%5BPUBMED-IDS%5D.
- ^ O'Malley JP, Maslen CL, Illingworth DR (19 May 1998). "Angiotensin-converting enzyme DD genotype and cardiovascular disease in heterozygous familial hypercholesterolemia". Circulation 97 (18): 1780–3. PMID 9603531. http://circ.ahajournals.org/cgi/content/full/97/18/1780.
- ^ Marks D, Wonderling D, Thorogood M, Lambert H, Humphries SE, Neil HA (June 2002). "Cost effectiveness analysis of different approaches of screening for familial hypercholesterolaemia". BMJ 324 (7349): 1303. doi:10.1136/bmj.324.7349.1303. PMC 113765. PMID 12039822. http://www.bmj.com/cgi/content/full/324/7349/1303. Full text at PMC: 113765
- ^ Umans-Eckenhausen MA, Defesche JC, Sijbrands EJ, Scheerder RL, Kastelein JJ (January 2001). "Review of first 5 years of screening for familial hypercholesterolaemia in the Netherlands". Lancet 357 (9251): 165–8. doi:10.1016/S0140-6736(00)03587-X. PMID 11213091.
- ^ Marks D, Thorogood M, Neil HA, Wonderling D, Humphries SE (March 2003). "Comparing costs and benefits over a 10 year period of strategies for familial hypercholesterolaemia screening" (PDF). J Public Health Med 25 (1): 47–52. doi:10.1093/pubmed/fdg010. PMID 12669918. http://jpubhealth.oxfordjournals.org/cgi/reprint/25/1/47.
- ^ National Institute for Health and Clinical Excellence. Clinical guideline 71: Familial hypercholesterolaemia. London, 2008.
- ^ Kastelein JJ, Akdim F, Stroes ES, et al. (April 2008). "Simvastatin with or without ezetimibe in familial hypercholesterolemia". N. Engl. J. Med. 358 (14): 1431–43. doi:10.1056/NEJMoa0800742. PMID 18376000.
- ^ Scientific Steering Committee on behalf of the Simon Broome Register Group (1999). "Mortality in treated heterozygous familial hypercholesterolaemia: implications for clinical management". Atherosclerosis 142 (1): 105–12. doi:10.1016/S0021-9150(98)00200-7. PMID 9920511.
- ^ Marais AD, Blom DJ, Firth JC (January 2002). "Statins in homozygous familial hypercholesterolemia". Curr Atheroscler Rep 4 (1): 19–25. doi:10.1007/s11883-002-0058-7. PMID 11772418.
- ^ Bilheimer DW, Goldstein JL, Grundy SM, Starzl TE, Brown MS (December 1984). "Liver transplantation to provide low-density-lipoprotein receptors and lower plasma cholesterol in a child with homozygous familial hypercholesterolemia". N. Engl. J. Med. 311 (26): 1658–64. doi:10.1056/NEJM198412273112603. PMID 6390206.
- ^ Revell SP, Noble-Jamieson G, Johnston P, Rasmussen A, Jamieson N, Barnes ND (November 1995). "Liver transplantation for homozygous familial hypercholesterolaemia". Arch. Dis. Child. 73 (5): 456–8. doi:10.1136/adc.73.5.456. PMC 1511367. PMID 8554367. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1511367. Full text at PMC: 1511367
- ^ López-Santamaria M, Migliazza L, Gamez M, et al. (April 2000). "Liver transplantation in patients with homozygotic familial hypercholesterolemia previously treated by end-to-side portocaval shunt and ileal bypass". J. Pediatr. Surg. 35 (4): 630–3. doi:10.1053/jpsu.2000.0350630. PMID 10770402. http://linkinghub.elsevier.com/retrieve/pii/S0022-3468(00)56892-4.
- ^ Buchwald H, Varco RL, Boen JR, et al. (June 1998). "Effective lipid modification by partial ileal bypass reduced long-term coronary heart disease mortality and morbidity: five-year posttrial follow-up report from the POSCH. Program on the Surgical Control of the Hyperlipidemias". Arch. Intern. Med. 158 (11): 1253–61. doi:10.1001/archinte.158.11.1253. PMID 9625405. http://archinte.ama-assn.org/cgi/content/full/158/11/1253.
- ^ Bilheimer DW, Goldstein JL, Grundy SM, Brown MS (December 1975). "Reduction in cholesterol and low density lipoprotein synthesis after portacaval shunt surgery in a patient with homozygous familial hypercholesterolemia". J. Clin. Invest. 56 (6): 1420–30. doi:10.1172/JCI108223. PMC 333120. PMID 172531. http://www.jci.org/articles/view/108223. Full text at PMC: 333120
- ^ Cuchel M, Bloedon LT, Szapary PO, et al. (January 2007). "Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia". N. Engl. J. Med. 356 (2): 148–56. doi:10.1056/NEJMoa061189. PMID 17215532. http://content.nejm.org/cgi/content/full/356/2/148.
- ^ Eriksson M, Carlson LA, Miettinen TA, Angelin B (August 1999). "Stimulation of fecal steroid excretion after infusion of recombinant proapolipoprotein A-I. Potential reverse cholesterol transport in humans". Circulation 100 (6): 594–8. PMID 10441095. http://circ.ahajournals.org/cgi/content/full/100/6/594.
- ^ Grossman M, Rader DJ, Muller DW, et al. (November 1995). "A pilot study of ex vivo gene therapy for homozygous familial hypercholesterolaemia". Nat. Med. 1 (11): 1148–54. doi:10.1038/nm1195-1148. PMID 7584986.
- ^ Mabuchi H, Koizumi J, Shimizu M, Takeda R (February 1989). "Development of coronary heart disease in familial hypercholesterolemia". Circulation 79 (2): 225–32. PMID 2914343.
- ^ a b Greene O, Durrington P (May 2004). "Clinical management of children and young adults with heterozygous familial hypercholesterolaemia in the UK". J R Soc Med 97 (5): 226–9. doi:10.1258/jrsm.97.5.226. PMC 1079462. PMID 15121812. http://jrsm.rsmjournals.com/cgi/content/full/97/5/226. Full text at PMC: 1079462
- ^ Rodenburg J, Vissers MN, Wiegman A, Trip MD, Bakker HD, Kastelein JJ (August 2004). "Familial hypercholesterolemia in children". Curr. Opin. Lipidol. 15 (4): 405–11. doi:10.1097/01.mol.0000137228.92396.f3. PMID 15243213.
- ^ Wiegman A, Hutten BA, de Groot E, et al. (July 2004). "Efficacy and safety of statin therapy in children with familial hypercholesterolemia: a randomized controlled trial". JAMA 292 (3): 331–7. doi:10.1001/jama.292.3.331. PMID 15265847. http://jama.ama-assn.org/cgi/content/full/292/3/331.
- ^ Kavey RE, Allada V, Daniels SR, et al. (December 2006). "Cardiovascular risk reduction in high-risk pediatric patients: a scientific statement from the American Heart Association Expert Panel on Population and Prevention Science; the Councils on Cardiovascular Disease in the Young, Epidemiology and Prevention, Nutrition, Physical Activity and Metabolism, High Blood Pressure Research, Cardiovascular Nursing, and the Kidney in Heart Disease; and the Interdisciplinary Working Group on Quality of Care and Outcomes Research: endorsed by the American Academy of Pediatrics". Circulation 114 (24): 2710–38. doi:10.1161/CIRCULATIONAHA.106.179568. PMID 17130340. http://circ.ahajournals.org/cgi/content/full/114/24/2710.
- ^ Müller C (1938). "Xanthoma, hypercholesterolemia, angina pectoris". Acta Med Scandinav 95 Suppl (89): 75. doi:10.1111/j.0954-6820.1938.tb19279.x.
- ^ Goldstein JL, Brown MS (October 1973). "Familial hypercholesterolemia: identification of a defect in the regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity associated with overproduction of cholesterol". Proc. Natl. Acad. Sci. U.S.A. 70 (10): 2804–8. doi:10.1073/pnas.70.10.2804. PMC 427113. PMID 4355366. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=427113.
- ^ Brown MS, Goldstein JL (January 1976). "Receptor-mediated control of cholesterol metabolism" (PDF). Science 191 (4223): 150–4. doi:10.1126/science.174194. PMID 174194. http://www.sciencemag.org/cgi/reprint/191/4223/150.
- ^ Nobelprize.org. "Medicine 1985". http://nobelprize.org/nobel_prizes/medicine/laureates/1985. Retrieved 2008-02-28.
External links
- MEDPED (Make Early Diagnosis to Prevent Early Deaths) - US screening program based at the University of Utah, Salt Lake City
- H·E·A·R·T UK - Hypercholesterolemia charity based in the United Kingdom
- Database of all known LDLR mutations (maintained by Leiden University Medical Centre, hosted by University College London)
- Familial hypercholesterolaemia - report of a WHO consultation - reproduction of WHO report WHO/HGN/FH/CONS/98.7 (1998) on the diagnosis and treatment of FH
Inborn error of lipid metabolism: dyslipidemia (E78, 272.0–272.6) Hyperlipidemia Hypercholesterolemia/Hypertriglyceridemia (Lipoprotein lipase deficiency/Type Ia, Familial apoprotein CII deficiency/Type Ib, Familial hypercholesterolemia/Type IIa, Combined hyperlipidemia/Type IIb, Familial dysbetalipoproteinemia/Type III, Familial hypertriglyceridemia/Type IV) · Xanthoma/XanthomatosisHypolipoproteinemia Lipodystrophy Other Categories:- Lipid metabolism disorders

Wikimedia Foundation. 2010.