- Abetalipoproteinemia
-
Abetalipoproteinemia Classification and external resources 
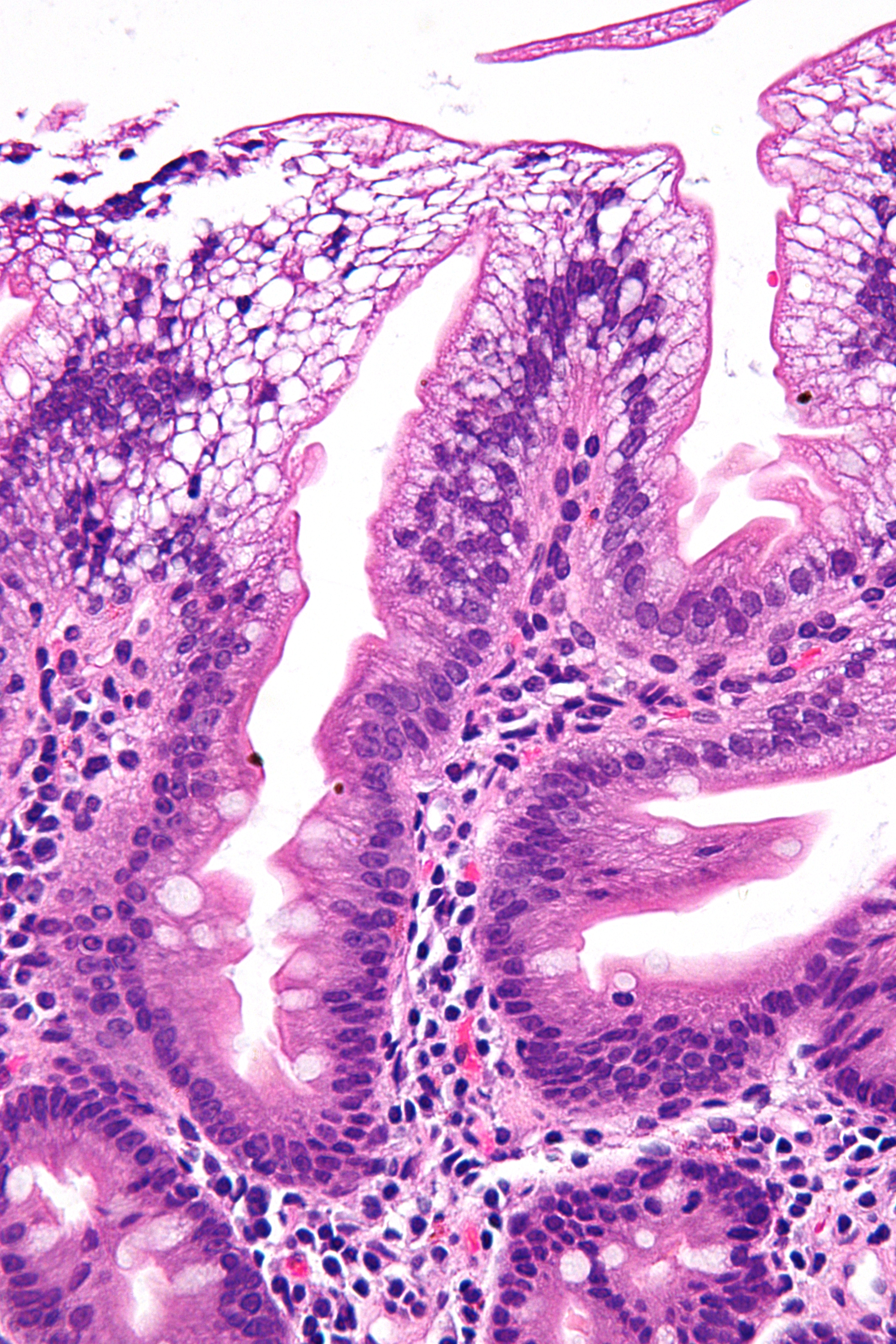
Micrograph showing enterocytes with a clear cytoplasm (due to lipid accumulation) characteristic of abetalipoproteinemia. Duodenal biopsy. H&E stain.ICD-10 E78.6 ICD-9 272.5 OMIM 200100 DiseasesDB 17 MedlinePlus 001666 eMedicine med/1117 MeSH D000012 Abetalipoproteinemia, or Bassen-Kornzweig syndrome,[1] is a rare autosomal recessive[2] disorder that interferes with the normal absorption of fat and fat-soluble vitamins from food. It is caused by a deficiency of apolipoprotein B-48 and B-100, which are used in the synthesis and exportation of chylomicrons and VLDL respectively. It is not to be confused with familial dysbetalipoproteinemia.
Contents
Features
Abetalipoproteinemia affects the absorption of dietary fats, cholesterol, and certain vitamins. People affected by this disorder are not able to make certain lipoproteins, which are molecules that consist of proteins combined with cholesterol and particular fats called triglycerides. This leads to a multiple vitamin deficiency, affecting the fat-soluble vitamin A, vitamin D, vitamin E, and vitamin K. However, many of the observed effects are due to vitamin E deficiency in particular.
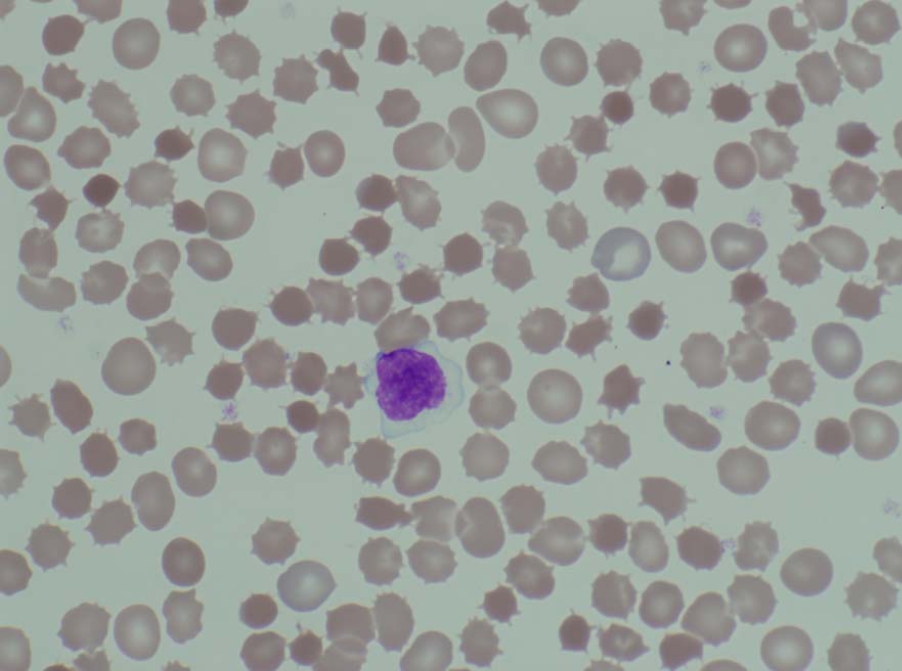
 Acanthocytosis in a patient with abetalipoproteinemia.
Acanthocytosis in a patient with abetalipoproteinemia.
The signs and symptoms of abetalipoproteinemia appear in the first few months of life(because pancreatic lipase is not active in this period). They can include failure to gain weight and grow at the expected rate (failure to thrive); diarrhea; abnormal star-shaped red blood cells (acanthocytosis); and fatty, foul-smelling stools (steatorrhea). The stool may contain large chunks of fat and/or blood. Other features of this disorder may develop later in childhood and often impair the function of the nervous system. They can include poor muscle coordination, difficulty with balance and movement (ataxia), and progressive degeneration of the light-sensitive layer (retina) at the back of the eye that can progress to near-blindness(due to deficiency of Vitamin A,retinol). Adults in their thirties or forties may have increasing difficulty with balance and walking. Many of the signs and symptoms of abetalipoproteinemia result from a severe vitamin deficiency, especially vitamin E deficiency, which typically results in eye problems with degeneration of the spinocerebellar and dorsal columns tracts.
Symptoms
Often symptoms will arise that indicate the body is not absorbing or making the lipoproteins that it needs. These symptoms usually appear en masse, meaning that they happen all together, all the time. These symptoms come as follows:
- Failure to grow in infancy
- Fatty, pale stools
- Frothy stools
- Foul smelling stools
- Protruding abdomen
- Mental retardation/developmental delay
- Dyspraxia, evident by age ten
- Muscle weakness
- Slurred speech
- Scoliosis (curvature of the spine)
- Progressive decreased vision
- Balance and coordination problems
- Retinitis Pigmentosa
Diagnosis
The inability to absorb fat in the ileum will result in steatorrhea, or fat in the stool. As a result, this can be clinically diagnosed when foul-smelling stool is encountered. Low levels of plasma chylomicron are also characteristic. Acanthocytes are seen on blood smear.
There is an absence of apolipoprotein B. On intestinal biopsy, vacuoles containing lipids are seen in enterocytes. Since there is no or little assimilation of chylomicrons, their levels in plasma remains low. This disorder may also result in fat accumulation in the liver (hepatic steatosis). Because the epithelial cells of the bowel lack the ability to place fats into chylomicrons, lipids accumulate at the surface of the cell, crowding the functions that are necessary for proper absorption.
Genetics

Mutations in the microsomal triglyceride transfer protein (MTTP) gene has been associated with this condition.[3] (Apolipoprotein B deficiency, a related condition, is associated with deficiencies of apolipoprotein B.)
The MTTP gene provides instructions for making a protein called microsomal triglyceride transfer protein, which is essential for creating beta-lipoproteins. These lipoproteins are both necessary for the absorption of fats, cholesterol, and fat-soluble vitamins from the diet and necessary for the efficient transport of these substances in the bloodstream. Most of the mutations in this gene lead to the production of an abnormally short microsomal triglyceride transfer protein, which prevents the normal creation of beta-lipoproteins in the body. MTTP-associated mutations are inherited in an autosomal recessive pattern, which means both copies of the gene must be faulty to produce the disease.
Treatment
Treatment normally consists of rigorous dieting, involving mass amounts of vitamin E. Vitamin E helps the body restore and produce lipoproteins, which people with abetalipoprotenimia usually lack. Vitamin E also helps keep skin and eyes healthy; studies show that many affected males will have vision problems later on in life. Dyspraxia and muscle weakness are usually treated with physiotherapy or occupational therapy. Dietary restriction of triglycerides has also been useful.
References
- ^ Bassen FA, Kornzweig AL (1950). "Malformation of the erythrocytes in a case of atypical retinitis pigmentosa". Blood 5 (4): 381–87. PMID 15411425. http://bloodjournal.hematologylibrary.org/cgi/reprint/5/4/381.
- ^ Benayoun, L; Granot, E; Rizel, L; Allon-Shalev, S; Behar, Dm; Ben-Yosef, T (April 2007). "Abetalipoproteinemia in Israel: evidence for a founder mutation in the Ashkenazi Jewish population and a contiguous gene deletion in an Arab patient". Molecular genetics and metabolism 90 (4): 453–7. doi:10.1016/j.ymgme.2006.12.010. PMID 17275380.
- ^ "Abetalipoproteinemia - Genetics Home Reference". http://ghr.nlm.nih.gov/condition=abetalipoproteinemia. Retrieved 2008-02-24.
External links
- Abetalipoproteinemia at NLM Genetics Home Reference
- AllRefer.com Bassen-Kornzweig syndrome
Inborn error of lipid metabolism: dyslipidemia (E78, 272.0–272.6) Hyperlipidemia Hypercholesterolemia/Hypertriglyceridemia (Lipoprotein lipase deficiency/Type Ia, Familial apoprotein CII deficiency/Type Ib, Familial hypercholesterolemia/Type IIa, Combined hyperlipidemia/Type IIb, Familial dysbetalipoproteinemia/Type III, Familial hypertriglyceridemia/Type IV) · Xanthoma/XanthomatosisHypolipoproteinemia Lipodystrophy Other Categories:- Autosomal recessive disorders
- Lipid metabolism disorders
- Rare diseases

Wikimedia Foundation. 2010.