- Hereditary hemorrhagic telangiectasia
-
Hereditary hemorrhagic telangiectasia Classification and external resources 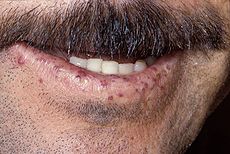
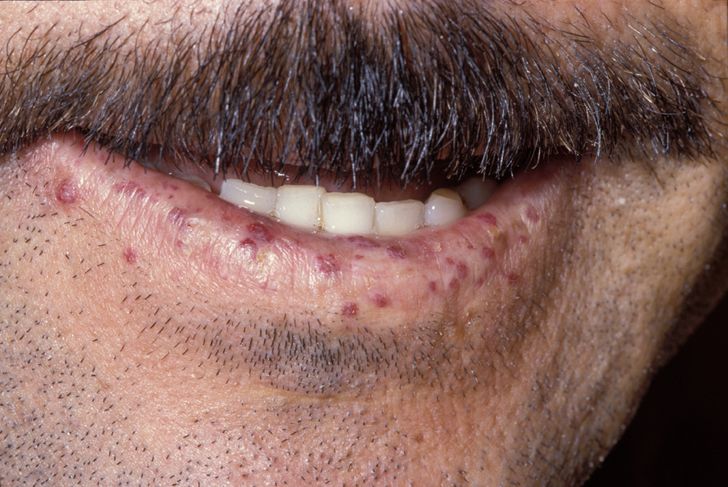
Characteristic lip telangiectases.ICD-10 I78.0 ICD-9 448.0 OMIM 187300 600376 601101 610655 175050 DiseasesDB 9303 eMedicine med/2764 ped/1668 derm/782 MeSH D013683 GeneReviews Hereditary Hemorrhagic Telangiectasia Hereditary hemorrhagic telangiectasia (HHT), also known as Osler-Weber-Rendu disease and Osler-Weber-Rendu syndrome, is a genetic disorder that leads to abnormal blood vessel formation in the skin, mucous membranes, and often in organs such as the lungs, liver and brain.[1][2]
It may lead to nosebleeds, acute and chronic digestive tract bleeding, and various problems due to the involvement of other organs. Treatment focuses on reducing bleeding from blood vessel lesions, and sometimes surgery or other targeted interventions to remove arteriovenous malformations in organs. Chronic bleeding often requires iron supplements and sometimes blood transfusions. HHT is transmitted in an autosomal dominant fashion, and occurs in one in 5,000 people.[1][2]
The disease carries the names of Sir William Osler, Henri Jules Louis Marie Rendu and Frederick Parkes Weber, who described it in the late 19th and early 20th centuries.[3]
Contents
Signs and symptoms
Telangiectasias
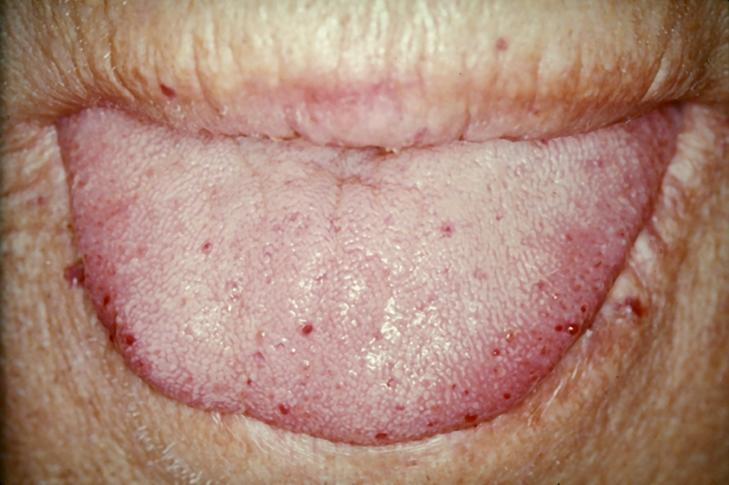
 Tongue telangiectases as seen in a person with hereditary hemorrhagic telangiectasia
Tongue telangiectases as seen in a person with hereditary hemorrhagic telangiectasia
Telangiectasia (small vascular malformations) may occur in the skin and mucosal linings of the nose and gastrointestinal tract. The most common problem is nosebleeds (epistaxis), which happen frequently from childhood and affect about 90–95% of people with HHT. Lesions on the skin and in the mouth bleed less often but may be considered cosmetically displeasing; they affect about 80%.[1][2] The skin lesions characteristically occur on the lips, the nose and the fingers, and on the skin of the face in sun-exposed areas. They appear suddenly, with the number increasing over time.[2]
A fifth are affected by symptomatic digestive tract lesions, although a higher number have lesions that are not causing symptoms. These lesions may bleed intermittently, which is rarely significant enough to be noticed (in the form of bloody vomiting or black stool), but eventually leads to depletion of iron in the body, resulting in iron-deficiency anemia.[1][2]
Arteriovenous malformation
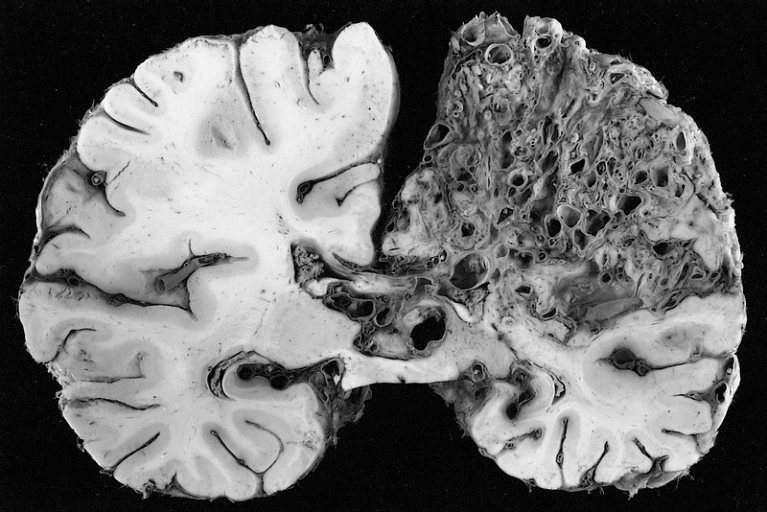
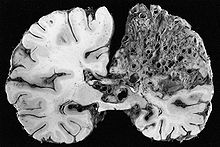 A very large arteriovenous malformation in the left hemisphere (on the right in this image) of the brain.
A very large arteriovenous malformation in the left hemisphere (on the right in this image) of the brain.Arteriovenous malformation (AVM, larger vascular malformations) occur in larger organs, predominantly the lungs (50%), liver (30–70%) and the brain (10%), with a very small proportion (<1%) having AVMs in the spinal cord.[1][2]
Vascular malformations in the lungs may cause a number of problems. The lungs normally "filter out" bacteria and blood clots from the bloodstream; AVMs bypass the capillary network of the lungs and allow these to migrate to the brain, where bacteria may cause a brain abscess and blood clots may lead to stroke.[1] HHT is the most common cause of lung AVMs: out of all people found to have lung AVMs, 70–80% are due to HHT.[4][5] Bleeding from lung AVMs is relatively unusual, but may cause hemoptysis (coughing up blood) or hemothorax (blood accumulating in the chest cavity).[1][2][4] Large vascular malformations in the lung allow oxygen-depleted blood from the right ventricle to bypass the alveoli, meaning that this blood does not have an opportunity absorb fresh oxygen. This may lead to breathlessness.[4][5] Large AVMs may lead to platypnea, difficulty in breathing more marked when sitting up compared to lying down; this probably reflects changes in blood flow associated with positioning.[4] Very large AVMs cause a marked inability to absorb oxygen, which may be noted by cyanosis (bluish discoloration of the lips and skin), clubbing of the fingernails (often encountered in chronically low oxygen levels), and a humming noise over the affected part of the lung detectable by stethoscope.[4][5]
The symptoms produced by AVMs in the liver depend on the type of abnormal connection that they form between blood vessels. If the connection is between arteries and veins, a large amount of blood bypasses the body's organs, for which the heart compensates by increasing the cardiac output. Eventually congestive cardiac failure develops ("high-output cardiac failure"), with breathlessness and leg swelling among other problems.[1][6] If the AVM creates a connection between the portal vein and the blood vessels of the liver, the result may be portal hypertension (increased portal vein pressure), in which collateral blood vessels form in the esophagus (esophageal varices), which may bleed violently; furthermore, the increased pressure may give rise to fluid accumulation in the abdominal cavity (ascites). If the flow in the AVM is in the other direction, portal venous blood flows directly into the veins rather than running through the liver; this may lead to hepatic encephalopathy (confusion due to portal waste products irritating the brain). Rarely, the bile ducts are deprived of blood, leading to severe cholangitis (inflammation of the bile ducts).[1][6] Liver AVMs are detectable in over 70% of people with HHT, but only 10% experience problems as a result.[2]
In the brain, AVMs occasionally exert pressure, leading to headaches. They may also increase the risk of seizures, as would any abnormal tissue in the brain. Finally, hemorrhage from an AVM may lead to intracerebral hemorrhage (bleeding into the brain), which causes any of the symptoms of stroke such as weakness in part of the body or difficulty speaking. If the bleeding occurs into the subarachnoid space (subarachnoid hemorrhage), there is usually a severe, sudden headache and decreased level of consciousness and often weakness in part of the body.[1][2]
Other problems
A very small proportion (those affected by SMAD4 mutations, see below) have multiple benign polyps in the large intestine, which may bleed or transform into colorectal cancer. A similarly small proportion experiences pulmonary hypertension, a state in which the pressure in the lung arteries is increased, exerting pressure on the right side of the heart and causing peripheral edema (swelling of the legs), fainting and attacks of chest pain. It has been observed that the risk of thrombosis (particularly venous thrombosis, in the form of deep vein thrombosis or pulmonary embolism) may be increased. There is a suspicion that those with HHT may have a mild immunodeficiency and are therefore at a slightly increased risk from infections.[1]
Genetics
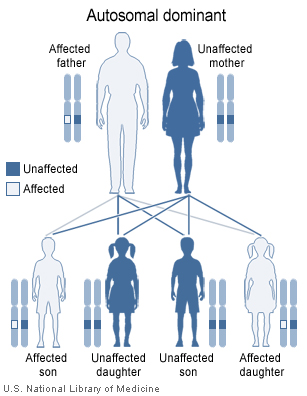
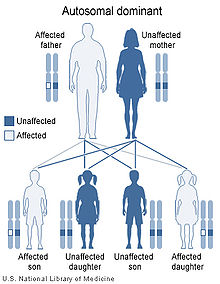 Hereditary hemorrhagic telangiectasia has an autosomal dominant pattern of inheritance.
Hereditary hemorrhagic telangiectasia has an autosomal dominant pattern of inheritance.HHT is a genetic disorder by definition. It is inherited in an autosomal dominant manner, which means that an affected person carries one abnormal gene with a 50% chance of passing this gene to offspring. Those with HHT symptoms that have no relatives with the disease may have a new mutation.[7] It seems that carrying two abnormal copies of the gene is not compatible with life, and hence no homozygotes have been described.[1]
Five genetic types of HHT are recognized. Of these, three have been linked to particular genes, while the two remaining have currently only been associated with a particular locus. More than 80% of all cases of HHT are due to mutations in either ENG or ACVRL1.[8] A total of over 600 different mutations is known. There is likely to be a predominance of either type in particular populations, but the data are conflicting. MADH4 mutations, which cause colonic polyposis in addition to HHT, comprise about 2% of disease-causing mutations. Apart from MADH4, it is not clear whether mutations in ENG and ACVRL1 lead to particular symptoms,[1] although some reports suggest that ENG mutations are more likely to cause lung problems while ACVRL1 mutations may cause more liver problems,[2][5] and pulmonary hypertension may be a particular problem in people with ACVRL1 mutations.[8] People with exactly the same mutations may have different nature and severity of symptoms, suggesting that additional genes or other risk factors may determine the rate at which lesions develop; these have not yet been identified.[2][8]
Name OMIM Gene Locus Description HHT1 187300 ENG 9q34.1 ENG codes for endoglin, a receptor of TGF-β1 (transforming growth factor beta 1) and TGF-β3; the genetic linkage was identified in 1994.[9] A high proportion of frameshift mutations has been observed.[1] Practically all mutations occur in the extracellular part of the protein (the part that sits on the surface of the cell).[8] HHT2 600376 ACVRL1 12q11-q14 ACVRL1 codes for Alk-1 (activin receptor-like kinase 1), a TGF-β1 receptor; genetic linkage was identified in 1996.[10] HHT3 601101 Unknown 5q31 Function unknown, linkage identified in 2005.[11] HHT4 610655 Unknown 7p14. Function unknown, linkage identified in 2006.[12] JPHT 175050 MADH4 18q21.1 MADH4 codes for SMAD4, an intracellular signalling protein for the TGF superfamily receptors. Mutations in this gene cause HHT and juvenile polyposis. Linkage was identified in 2004.[13] Mutations mostly in exons 8–11, often de novo (newly acquired, not inherited).[1] Pathophysiology
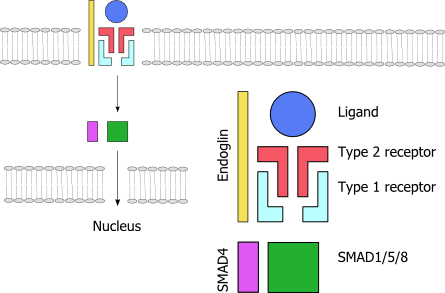
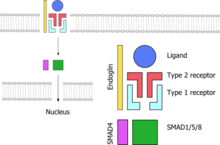 A schematic representation of the TGF-β signaling pathway. Endoglin (yellow) is needed for signalling. The ligand (blue) binds to the receptor complex; red indicates a type II receptor protein, which activates a type I receptor protein (turquiose) such as alk-1, which in turn phosphorylates a SMAD-based nuclear transcription factor (green and purple).
A schematic representation of the TGF-β signaling pathway. Endoglin (yellow) is needed for signalling. The ligand (blue) binds to the receptor complex; red indicates a type II receptor protein, which activates a type I receptor protein (turquiose) such as alk-1, which in turn phosphorylates a SMAD-based nuclear transcription factor (green and purple).Telangiectasias and arteriovenous malformations in HHT are thought to arise because of changes in angiogenesis, the development of blood vessels out of existing ones. The development of a new blood vessels requires the activation and migration of various types of cell, chiefly endothelium, smooth muscle and pericytes. The exact mechanism by which the HHT mutations influence this process is not yet clear, and it is likely that they disrupt a balance between pro- and antiangiogenic signals in blood vessels. The wall of telangiectasias is unusually friable, which explains the tendency of these lesions to bleed.[1]
All genes known so far to be linked to HHT code for proteins in the TGF-β signaling pathway. This is a group of proteins that participates in signal transduction of hormones of the transforming growth factor beta superfamily (the transforming growth factor beta, bone morphogenetic protein and growth differentiation factor classes), specifically BMP9/GDF2 and BMP10. The hormones do not enter the cell but link to receptors on the cell membrane; these then activate other proteins, eventually influencing cellular behavior in a number of ways such as cellular survival, proliferation (increasing in number) and differentiation (becoming more specialized).[1] For the hormone signal to be adequately transduced, a combination of proteins is needed: two each of two types of serine/threonine-specific kinase type membrane receptors and endoglin. When bound to the hormone, the type II receptor proteins phosphorylate (transfer phosphate) onto type I receptor proteins (of which Alk-1 is one), which in turn phosphorylate a complex of SMAD proteins (chiefly SMAD1, SMAD5 and SMAD8). These bind to SMAD4 and migrate to the cell nucleus where they act as transcription factors and participate in the transcription of particular genes. In addition to the SMAD pathway, the membrane receptors also act on the MAPK pathway, which has additional actions on the behavior of cells.[2] Both Alk-1 and endoglin are expressed predominantly in endothelium, perhaps explaining why HHT-causing mutations in these proteins lead predominantly to blood vessel problems.[2][8] Both ENG and ACVRL1 mutations lead predominantly to underproduction of the related proteins, rather than misfunctioning of the proteins.[8]
Diagnosis
Diagnostic tests may be conducted for various reasons. Firstly, some tests are needed to confirm or refute the diagnosis. Secondly, some are needed to identify any potential complications.[7]
Telangiectasias
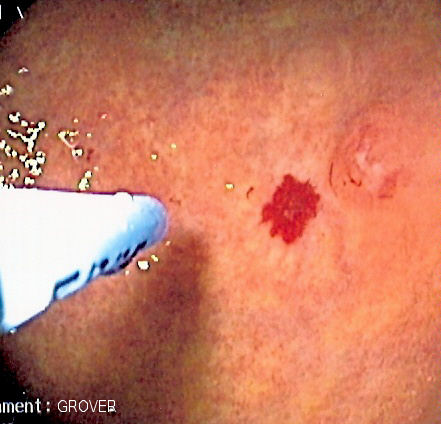
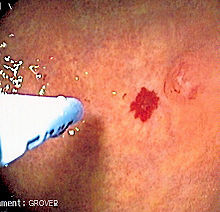 A vascular lesion in the digestive tract, being treated with argon plasma coagulation.
A vascular lesion in the digestive tract, being treated with argon plasma coagulation.The skin and oral cavity telangiectasias are visually identifiable on physical examination, and similarly the lesions in the nose may be seen on endoscopy of the nasopharynx or on laryngoscopy. The severity of nosebleeds may be quantified objectively using a grid-like questionnaire in which the number of nosebleed episodes and their duration is recorded.[2]
Digestive tract telangiectasias may be identified on esophagogastroduodenoscopy (endoscopy of the esophagus, stomach and first part of the small intestine). This procedure will typically only be undertaken if there is anemia that is more marked than expected by the severity of nosebleeds, or if there is evidence of severe bleeding (vomiting blood, black stools). If the number of lesions seen on endoscopy is unexpectedly low, the remainder of the small intestine may be examined with capsule endoscopy, in which the patient swallows a capsule-shaped device containing a miniature camera which transmits images of the digestive tract to a portable digital recorder.[2]
Arteriovenous malformations
Identification of AVMs requires detailed medical imaging of the organs most commonly affected by these lesions. Not all AVMs cause symptoms or are at risk of doing so, and hence there is a degree of variation between specialists as to whether such investigations would be performed, and by which modality; often, decisions on this issue are reached together with the patient.[1]
Lung AVMs may be suspected because of the abnormal appearance of the lungs on a chest X-ray, or hypoxia (low oxygen levels) on pulse oximetry or arterial blood gas determination. Bubble contrast echocardiography (bubble echo) may be used as a screening tool to identify abnormal connections between the lung arteries and veins. This involves the injection of agitated saline into a vein, followed by ultrasound-based imaging of the heart. Normally, the lungs remove small air bubbles from the circulation, and they are therefore only seen in the right atrium and the right ventricle. If an AVM is present, bubbles appear in the left atrium and left ventricle, usually 3–10 cardiac cycles after the right side; this is slower than in heart defects, in which there are direct connections between the right and left side of the heart. A larger number of bubbles is more likely to indicate the presence of an AVM. Bubble echo is not a perfect screening tool as it can miss smaller AVMs and does not identify the site of AVMs. Often contrast-enhanced computed tomography (CT angiography) is used to identify lung lesions; this modality has a sensitivity of over 90%.[1][2] It may be possible to omit contrast administration on modern CT scanners.[5] Echocardiography is also used if there is a suspicion of pulmonary hypertension or high-output cardiac failure due to large liver lesions, sometimes followed by cardiac catheterization to measure the pressures inside the various chambers of the heart.[1]
Liver AVMs may be suspected because of abnormal liver function tests in the blood, because the symptoms of heart failure develop, or because of jaundice or other symptoms of liver dysfunction. The most reliable initial screening test is Doppler ultrasonography of the liver; this has a very high sensitivity for identifying vascular lesions in the liver. If necessary, contrast-enhanced CT may be used to further characterize AVMs.[1][2][6] It is extremely common to find incidental nodules on liver scans, most commonly due to focal nodular hyperplasia (FNH), as these are a hundredfold times more common in HHT compared to the general population. FNH is regarded as harmless. Generally, tumor markers and additional imaging modalities are used to differentiate between FNH and malignant tumors of the liver. Liver biopsy is discouraged in people with HHT as the risk of hemorrhage from liver AVMs may be significant.[6][7] Liver scans may be useful if someone is suspected of HHT, but does not meet the criteria (see below) unless liver lesions can be demonstrated.[7]
Brain AVMs may be detected on computed tomography angiography (CTA or CT angio) or magnetic resonance angiography (MRA); CTA is better in showing the vessels themselves, and MRA provides more detail about the relationship between an AVM and surrounding brain tissue.[14] In general, MRI is recommended.[2][7] Various types of vascular malformations may be encountered: AVMs, micro-AVMs, telangiectasias and arteriovenous fistulas.[7] If surgery, embolization, or other treatment is contemplated (see below), cerebral angiography may be required to get sufficient detail of the vessels. This procedure carries a small risk of stroke (0.5%) and is therefore limited to specific circumstances.[7][14] Recent professional guidelines recommend that all children with suspected or definite HHT undergo a brain MRI early in life to identify AVMs that can cause major complications.[7] Others suggest that screening for cerebral AVMs is probably unnecessary in those who are not experiencing any neurological symptoms, because most lesions discovered on screening scans would not require treatment, creating undesirable conundrums.[1]
Genetic testing
Genetic tests are available for the ENG, ACVRL1 and MADH4 mutations. Testing is not always needed for diagnosis, because the symptoms are sufficient to distinguish the disease from other diagnoses. There are situations in which testing can be particularly useful. Firstly, children and young adults with a parent with definite HHT may have limited symptoms, yet be at risk from some of the complications mentioned above; if the mutation is known in the affected parent, absence of this mutation in the child would prevent the need for screening tests. Furthermore, genetic testing may confirm the diagnosis in those with limited symptoms who otherwise would have been labeled "possible HHT" (see below).[7]
Genetic diagnosis in HHT is difficult, as mutations occur in numerous different locations in the linked genes, without particular mutations being highly frequent (as opposed to, for instance, the ΔF508 mutation in cystic fibrosis). Sequence analysis of the involved genes is therefore the most useful approach (sensitivity 75%), followed by additional testing to detect large deletions and duplications (additional 10%). Not all mutations in these genes have been linked with disease.[7]
Mutations in the MADH4 gene is usually associated with juvenile polyposis, and detection of such a mutation would indicate a need to screen the patient and affected relatives for polyps and tumors of the large intestine.[7]
Criteria
The diagnosis of made depending on the presence of four criteria, known as the "Curaçao criteria".[15] If three or four are met, a patient has "definite HHT", while two gives "possible HHT":
- Spontaneous recurrent epistaxis
- Multiple telangiectasias in typical locations (see above)
- Proven visceral AVM (lung, liver, brain, spine)
- First-degree family member with HHT
Despite the designation "possible", someone with a visceral AVM and a family history but no nosebleeds or telangiectasias is still extremely likely to have HHT, because these AVMs are very uncommon in the general population. At the same time, the same cannot be said of nosebleeds and sparse telangiectasias, both of which occur in people without HHT, in the absence of AVMs. Someone's diagnostic status may change in the course of life, as young children may not yet exhibit all the symptoms; at age 16, thirteen percent are still indeterminate, while at age 60 the vast majority (99%) have a definite diagnostic classification. The children of established HHT patients may therefore be labeled as "possible HHT", as 50% may turn out to have HHT in the course of their life.[1]
Treatment
Treatment of HHT is symptomatic (it deals with the symptoms rather than the disease itself), as there is no therapy that stops the development of telangiectasias and AVMs directly. Furthermore, some treatments are applied to prevent the development of common complications.[7] Chronic nosebleeds and digestive tract bleeding can both lead to anemia; if the bleeding itself cannot be completely stopped, the anemia requires treatment with iron supplements. Those who cannot tolerate iron tablets or solutions may require administration of intravenous iron sucrose, and blood transfusion if the anemia is causing severe symptoms that warrant rapid improvement of the blood count.[2][7]
Most treatments used in HHT have been described in adults, and the experience in treating children is more limited.[7] Women with HHT who get pregnant are at an increased risk of complications, and are observed closely, although the absolute risk is still low (1%).[1]
Nosebleeds
An acute nosebleed may be managed with a variety of measures, such as packing of the nasal cavity with absorbent swabs or gels. Removal of the packs after the bleeding may lead to reopening of the fragile vessels, and therefore lubricated or atraumatic packing is recommended.[7] Some patients may wish to learn packing themselves to deal with nosebleeds without having to resort to medical help.[16]
Frequent nosebleeds can be prevented in part by keeping the nostrils moist, and by applying saline solution, estrogen-containing creams or tranexamic acid; these have few side effects and may have a small degree of benefit.[7] A number of additional modalities has been used to prevent recurrent bleeding if simple measures are unsuccessful. Medical therapies include oral tranexamic acid and estrogen; the evidence for these is relatively limited, and estrogen is poorly tolerated by men and possibly carries risks of cancer and heart disease in women past the menopause.[2][7] Nasal coagulation and cauterization may reduce the bleeding from telangiectasias, and is recommended before surgery is considered; often, several sessions are needed. It may be possible to embolize vascular lesions through interventional radiology; this requires passing a catheter through a large artery and locating the maxillary artery under X-ray guidance, followed by the injection into the vessel of particles that occlude the blood vessels. The benefit from the procedure tends to be short-lived,[7] and it may be most appropriate in episodes of severe bleeding.[16]
If other interventions have failed, several operations have been reported to provide benefit. One is septal dermoplasty or Saunders' procedure,[17] in which skin is transplanted into the nostrils, and the other is Young's procedure,[18] in which the nostrils are sealed off completely.[7][16]
Skin and digestive tract
The skin lesions of HHT can be disfiguring, and may respond to treatment with long-pulsed Nd:YAG laser.[2] Skin lesions in the fingertips may sometimes bleed and cause pain. Skin grafting is occasionally needed to treat this problem.[2]
With regards to digestive tract lesions, mild bleeding and mild resultant anemia is treated with iron supplementation, and no specific treatment is administered. There is limited data on hormone treatment and tranexamic acid to reduce bleeding and anemia. Severe anemia or episodes of severe bleeding are treated with endoscopic argon plasma coagulation (APC) or laser treatment of any lesions identified; this may reduce the need for supportive treatment. The expected benefits are not such that repeated attempts at treating lesions are advocated.[7] Sudden, very severe bleeding is unusual—if encountered, alternative causes (such as a peptic ulcer) need to be considered[7]—but embolization may be used in such instances.[1]
Lung AVMs
Lung lesions, once identified, are usually treated to prevent episodes of bleeding and more importantly embolism to the brain. This is particularly done in lesions with a feeding blood vessel of 3 mm or larger, as these are the most likely to cause long-term complications unless treated. The most effective current therapy is embolization with detachable metal coils. The procedure involves puncture of a large vein (usually under a general anesthetic), followed by advancing of a catheter through the right ventricle and into the pulmonary artery, after which radiocontrast is injected to visualize the AVMs (pulmonary angiography). Once the lesion has been identified, coils are deployed that obstruct the blood flow and allow the lesion to regress. In experienced hands, the procedure tends to be very effective and with limited side effects, but lesions may recur and further attempts may be required. CTA scans are repeated to monitor for recurrence.[2][4][5][7] Surgical excision has now essentially been abandoned due to the success of embolotherapy.[5][7]
Those with either definite pulmonary AVMs or an abnormal contrast echocardiogram with no clearly visible lesions are deemed to be at risk from brain emboli. They are therefore counselled to avoid scuba diving, during which small air bubbles may form in the bloodsteam that may migrate to the brain and cause stroke. Similarly, antimicrobial prophylaxis is advised during procedures in which bacteria may enter the bloodstream, such as dental work, and avoidance of air bubbles during intravenous therapy.[2][5][7]
Liver AVMs
Given that liver AVMs generally cause high-output cardiac failure, the emphasis is on treating this with diuretics to reduce the circulating blood volume, restriction of salt and fluid intake, and antiarrhythmic agents in case of irregular heart beat. This may be sufficient in treating the symptoms of swelling and breathlessness. If this treatment is not effective or leads to side effects or complications, the only remaining option is liver transplantation. This is reserved for those with severe symptoms, as it carries a mortality of about 10%, but leads to good results if successful.[6][7] The exact point at which liver transplantion is to be offered is not yet completely established.[6] Embolization treatment has been attempted, but leads to severe complications in a proportion of patients and is discouraged.[5][6][7]
Other liver-related complications (portal hypertension, esophageal varices, ascites, hepatic encephalopathy) are treated with the same modalities as used in cirrhosis, although the use of transjugular intrahepatic portosystemic shunt treatment is discouraged due to the lack of documented benefit.[6]
Brain AVMs
The decision to treat brain AVMs depends on the symptoms that they cause (such as seizures or headaches). The bleeding risk is predicted by previous episodes of hemorrhage, and whether on the CTA or MRA scan the AVM appears to be deep seated or have deep venous drainage. Size of the AVM and the presence of aneurysms appears to matter less.[14] In HHT, some lesions (high-flow arteriovenous fistulae) tend to cause more problems, and treatment is warranted. Other AVMs may regress over time without intervention.[7] Various modalities are available, depending on the location of the AVM and its size: surgery, radiation-based treatment and embolization. Sometimes, multiple modalities are used on the same lesion.[2][14]
Surgery (by craniotomy, open brain surgery) may be offered based on the risks of treatment as determined by the Spetzler–Martin scale (grade I-V); this score is higher in larger lesions that are close to important brain structures and have deep venous drainage. High grade lesions (IV and V) have an unacceptably high risk and surgery is not typically offered in those cases. Radiosurgery (using targeted radiation therapy such as by a gamma knife) may be used if the lesion is small but close to vital structures. Finally, embolization may be used on small lesions that have only a single feeding vessel.[14]
Epidemiology
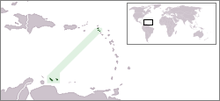 The Netherlands Antilles, where HHT is more common than anywhere in the world, located off the coast of Venezuela.
The Netherlands Antilles, where HHT is more common than anywhere in the world, located off the coast of Venezuela.Population studies from numerous areas in the world have shown that HHT occurs at roughly the same rate in almost all populations: somewhere around 1 in 5000. In some areas, it is much more common; for instance, in the French region of Haut Jura the rate is 1:2351 - twice as common as in other populations. This has been attributed to a founder effect, in which a population descending from a small number of ancestors has a high rate of a particular genetic trait because one of these ancestors harbored this trait.[8] In Haut Jura, this has been shown to be the result of a particular ACVRL1 mutation (named c.1112dupG or c.1112_1113insG).[2] The highest rate of HHT is 1:1331, reported in Bonaire and Curaçao, two islands in the Caribbean belonging to the Netherlands Antilles.[8]
Most people with HHT have a normal lifespan.[1] The skin lesions and nosebleeds tend to develop during childhood. AVMs are probably present from birth, but don't necessarily cause any symptoms. Frequent nosebleeds are the most common symptom and can significantly affect quality of life.[7]
History
Several 19th century English physicians, starting with Henry Gawen Sutton (1836–1891)[19] and followed by Benjamin Guy Babington (1794–1866)[20] and John Wickham Legg (1843–1921),[21] described the most common features of HHT, particularly the recurrent nosebleeds and the hereditary nature of the disease. The French physician Henri Jules Louis Marie Rendu (1844–1902) observed the skin and mucosal lesions, and distinguished the condition from hemophilia.[22] The Canadian-born Sir William Osler (1849–1919), then at Johns Hopkins Hospital and later at Oxford University, made further contributions with a 1901 report in which he described characteristic lesions in the digestive tract.[23] The English physician Frederick Parkes Weber (1863–1962) reported further on the condition in 1907 with a series of cases.[24] The term "hereditary hemorrhagic telangiectasia" was first used by the American physician Frederic M. Hanes (1883–1946) in a 1909 article on the condition.[3][25]
The diagnosis of HHT remained a clinical one until the genetic defects that cause HHT were identified by a research group at Duke University Medical Center, in 1994 and 1996 respectively.[9][10] In 2000, the international scientific advisory committee of HHT Foundation International published the now widely used Curaçao criteria.[7][15] In 2006, a group of international experts met in Canada and formulated an evidence-based guideline, sponsored by the HHT Foundation International.[7]
References
- ^ a b c d e f g h i j k l m n o p q r s t u v w x y z Govani FS, Shovlin CL (July 2009). "Hereditary haemorrhagic telangiectasia: a clinical and scientific review". Eur. J. Hum. Genet. 17 (7): 860–71. doi:10.1038/ejhg.2009.35. PMC 2986493. PMID 19337313. http://www.nature.com/ejhg/journal/v17/n7/full/ejhg200935a.html.
- ^ a b c d e f g h i j k l m n o p q r s t u v w x y z Dupuis-Girod S, Bailly S, Plauchu H (March 2010). "Hereditary hemorrhagic telangiectasia (HHT): from molecular biology to patient care". J. Thromb. Haemost. 8 (7): 1447–56. doi:10.1111/j.1538-7836.2010.03860.x. PMID 20345718.
- ^ a b Fuchizaki U, Miyamori H, Kitagawa S, Kaneko S, Kobayashi K (November 2003). "Hereditary haemorrhagic telangiectasia (Rendu-Osler-Weber disease)". Lancet 362 (9394): 1490–4. doi:10.1016/S0140-6736(03)14696-X. PMID 14602446.
- ^ a b c d e f Gossage JR, Kanj G (August 1998). "Pulmonary arteriovenous malformations. A state of the art review". Am. J. Respir. Crit. Care Med. 158 (2): 643–61. PMID 9700146. http://ajrccm.atsjournals.org/cgi/content/full/158/2/643.
- ^ a b c d e f g h i Faughnan ME, Granton JT, Young LH (May 2009). "The pulmonary vascular complications of hereditary haemorrhagic telangiectasia". Eur. Respir. J. 33 (5): 1186–94. doi:10.1183/09031936.00061308. PMID 19407052.
- ^ a b c d e f g h Buscarini E, Plauchu H, Garcia Tsao G, et al. (November 2006). "Liver involvement in hereditary hemorrhagic telangiectasia: consensus recommendations". Liver Int. 26 (9): 1040–6. doi:10.1111/j.1478-3231.2006.01340.x. PMID 17032403.
- ^ a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad Faughnan ME, Palda VA, Garcia-Tsao G, et al. (2011). "International guidelines for the diagnosis and management of hereditary hemorrhagic telangiectasia". J. Med. Genet. 48 (2): 73–87. doi:10.1136/jmg.2009.069013. PMID 19553198.
- ^ a b c d e f g h Abdalla SA, Letarte M (February 2006). "Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease". J. Med. Genet. 43 (2): 97–110. doi:10.1136/jmg.2005.030833. PMC 2603035. PMID 15879500. http://jmg.bmj.com/content/43/2/97.long.
- ^ a b McAllister KA, Grogg KM, Johnson DW, et al. (December 1994). "Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1". Nat. Genet. 8 (4): 345–51. doi:10.1038/ng1294-345. PMID 7894484.
- ^ a b Johnson DW, Berg JN, Baldwin MA, et al. (June 1996). "Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2". Nat. Genet. 13 (2): 189–95. doi:10.1038/ng0696-189. PMID 8640225.
- ^ Cole SG, Begbie ME, Wallace GM, Shovlin CL (2005). "A new locus for hereditary haemorrhagic telangiectasia (HHT3) maps to chromosome 5". J. Med. Genet. 42 (7): 577–82. doi:10.1136/jmg.2004.028712. PMC 1736109. PMID 15994879. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1736109.
- ^ Bayrak-Toydemir P, McDonald J, Akarsu N, et al. (2006). "A fourth locus for hereditary hemorrhagic telangiectasia maps to chromosome 7". Am. J. Med. Genet. A 140 (20): 2155–62. doi:10.1002/ajmg.a.31450. PMID 16969873.
- ^ Gallione CJ, Repetto GM, Legius E, et al. (March 2004). "A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4)". Lancet 363 (9412): 852–9. doi:10.1016/S0140-6736(04)15732-2. PMID 15031030.
- ^ a b c d e Friedlander RM (June 2007). "Clinical practice. Arteriovenous malformations of the brain". N. Engl. J. Med. 356 (26): 2704–12. doi:10.1056/NEJMcp067192. PMID 17596605.
- ^ a b Shovlin CL, Guttmacher AE, Buscarini E, et al. (Mar 2000). "Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome)". Am. J. Med. Genet. 91 (1): 66–7. doi:10.1002/(SICI)1096-8628(20000306)91:1<66::AID-AJMG12>3.0.CO;2-P. PMID 10751092.
- ^ a b c Geisthoff UW, Fiorella ML, Fiorella R (2006). "Treatment of recurrent epistaxis in HHT". Curr. Pharm. Des. 12 (10): 1237–42. doi:10.2174/138161206776361255. PMID 16611115.
- ^ Saunders WH (1963). "Septal dermoplasty: a new operative procedure for control of nosebleeds in patients with hereditary hæmorrhagic telangiectasia". J. Laryngol. Otol. 77 (01): 69–76. doi:10.1017/S0022215100060382. PMID 13986828.
- ^ Young A (May 1967). "Closure of the nostrils in atrophic rhinitis". J. Laryngol. Otol. 81 (5): 515–24. doi:10.1017/S0022215100067426. PMID 6024992.
- ^ Sutton HG (1864). "Epistaxis as an indication of impaired nutrition, and of degeneration of the vascular system". Med. Mirror: 769–81.
- ^ Babington BG (1865). "Hereditary epistaxis". Lancet 2 (2195): 362–363. doi:10.1016/S0140-6736(02)55197-7.
- ^ Legg W (1876). "A case of haemophilia complicated with multiple naevi". Lancet 2 (2781): 856–7. doi:10.1016/S0140-6736(02)49594-3.
- ^ Rendu HJ (1896). "Épistaxis répétées chez un sujet porteur de petits angiomes cutanés et muqueux". Gaz. Hop.: 1322–3.
- ^ Osler W (1901). "On a family form of recurring epistaxis, associated with multiple telangiectases of the skin and mucous membranes". Bull. Johns Hopkins Hosp. 12: 333–7.
- ^ Weber FP (1907). "Multiple hereditary developmental angiomata (telangiectases) of the skin and mucous membranes associated with recurring haemorrhages". Lancet 2 (4377): 160–162. doi:10.1016/S0140-6736(00)32590-9.
- ^ Hanes FM (1909). "Multiple hereditary telangiectasis causing hemorrhage (hereditary hemorrhagic telangiectasia)". Bull. Johns Hopkins Hosp. 20: 63–73.
External links
- HHT Foundation International
- HHT mutation database (ENG and ACVRL1)
Genetic disorder, membrane: cell surface receptor deficiencies G protein-coupled receptor
(including hormone)Class ATSHR (Congenital hypothyroidism 1) · LHCGR (Male-limited precocious puberty) · FSHR (XX gonadal dysgenesis) · EDNRB (ABCD syndrome, Waardenburg syndrome 4a, Hirschsprung's disease 2) · AVPR2 (Nephrogenic diabetes insipidus 1) · PTGER2 (Aspirin-induced asthma)Class BClass CCASR (Familial hypocalciuric hypercalcemia)Class FFZD4 (Familial exudative vitreoretinopathy 1)Enzyme-linked receptor
(including
growth factor)ROR2 (Robinow syndrome) · FGFR1 (Pfeiffer syndrome, KAL2 Kallmann syndrome) · FGFR2 (Apert syndrome, Antley-Bixler syndrome, Pfeiffer syndrome, Crouzon syndrome, Jackson-Weiss syndrome) · FGFR3 (Achondroplasia, Hypochondroplasia, Thanatophoric dysplasia, Muenke syndrome) · INSR (Donohue syndrome · Rabson–Mendenhall syndrome) · NTRK1 (Congenital insensitivity to pain with anhidrosis) · KIT (KIT Piebaldism, Gastrointestinal stromal tumor)AMHR2 (Persistent Mullerian duct syndrome II)
TGF beta receptors: Endoglin/Alk-1/SMAD4 (Hereditary hemorrhagic telangiectasia) · TGFBR1/TGFBR2 (Loeys-Dietz syndrome)JAK-STAT TNF receptor Lipid receptor LRP: LRP2 (Donnai-Barrow syndrome) · LRP4 (Cenani Lenz syndactylism) · LRP5 (Worth syndrome, Familial exudative vitreoretinopathy 4, Osteopetrosis 1)
LDLR (LDLR Familial hypercholesterolemia)Other/ungrouped Immunoglobulin superfamily: AGM3, 6
Integrin: LAD1 · Glanzmann's thrombasthenia · Junctional epidermolysis bullosa with pyloric atresia
EDAR (EDAR Hypohidrotic ectodermal dysplasia) · PTCH1 (Nevoid basal cell carcinoma syndrome) · BMPR1A (BMPR1A Juvenile polyposis syndrome) · IL2RG (X-linked severe combined immunodeficiency)Categories:- Diseases of arteries, arterioles and capillaries
- Vascular-related cutaneous conditions
- Respiratory diseases
- Neurological disorders
- Autosomal dominant disorders
- Cell surface receptor deficiencies
Wikimedia Foundation. 2010.