- Myotonic dystrophy
-
Myotonic dystrophy Classification and external resources ICD-10 G71.1 OMIM 160900 602668 DiseasesDB 8739 MeSH D009223 Myotonic dystrophy (dystrophia myotonica, DM) is a chronic, slowly progressing, highly variable inherited multisystemic disease. It is characterized by wasting of the muscles (muscular dystrophy), cataracts, heart conduction defects, endocrine changes, and myotonia. Myotonic dystrophy can occur in patients of any age. Genetically the disease is autosomal dominant.
Contents
Classification
Comparison between myotonic dystrophy subtypes Type Gene Repeat Anticipation Severity DM1 DMPK CTG Yes Moderate-severe DM2 ZNF9 CCTG Minimal/none Mild-moderate Two types of adult onset myotonic dystrophy exist. Type 1 (DM1), also called Steinert's disease, has a severe congenital form and a milder childhood-onset form. Myotonic dystrophy type 2 (DM2), also called proximal myotonic myopathy (PROMM), is due to a different mechanism than DM1 and generally manifests with milder signs and symptoms.
Other forms of myotonic dystrophy (DM3, DM4, and DMX) may also exist.[citation needed] One recent case was proposed as a candidate for the "DM3" label,[1] but was later characterized as a form of Paget's disease.[2][3]
Epidemiology
Myotonic dystrophy is the most common form of muscular dystrophy diagnosed in adults.[citation needed] Myotonic dystrophy affects at least 1 in 8,000 people worldwide. The prevalence of the two types of myotonic dystrophy varies among different geographic and ethnic populations. In most populations, type 1 appears to be more common than type 2. However, recent studies suggest that type 2 may be as common as type 1 among people in Germany and Finland.[4]
Symptoms and signs
 40-year-old patient with myotonic dystrophy presenting with bilateral cataracts and complete heart block.
40-year-old patient with myotonic dystrophy presenting with bilateral cataracts and complete heart block.
Presentation of symptoms and signs varies considerably by form (DM1/DM2), severity and even unusual DM2 phenotypes. DM1 patients often present with myotonia, disabling distal weakness and severe cognitive problems. DM2 patients commonly present with muscle pain, stiffness, fatigue, or the development of proximal lower extremity weakness (Day et al, 2003). The characteristic pattern of weakness is different for DM1 and DM2: In DM1, it is noted in face and jaw muscles, the drooping of the eyelids (ptosis), weakness of the neck muscles, hands and lower legs. In DM2, the weakness is more evident in proximal muscles, those closer to the trunk of the body: neck, shoulders, hip flexors and upper legs.
Symptoms and signs classically associated with DM1 are generally more mild and involve the smooth muscle (including G.I. symptoms), hypersomnia (daytime sleepiness), muscle wasting, dysphagia, and respiratory insufficiency. In addition, DM1 may manifest with a cognitive abnormalities including developmental delays, learning problems, language, speech, behaviour, apathy or hypersomnia. Cognitive manifestations for DM2 include problems with executive function (e.g., organization, concentration, word-finding) and hypersomnia. Conduction abnormalities are more common in DM1 than DM2, but all patients are advised to have an annual ECG. Both types are also associate with insulin resistance.
DM2 is generally milder than DM1, with generally fewer DM2 patients requiring assistive devices than DM1 patients.[citation needed] In addition, the severe congenital form that affects babies in DM1 has not been found in DM2 and the early onset of symptoms is rarely noted to appear in younger patients in the medical literature.
Genetics
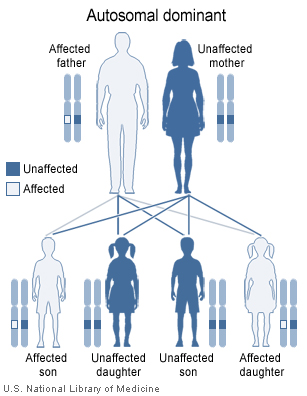
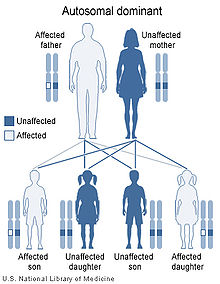 Myotonic dystrophy is inherited in an autosomal dominant pattern.
Myotonic dystrophy is inherited in an autosomal dominant pattern.Myotonic dystrophy is a genetic condition which is inherited in an autosomal dominant pattern and thus will be passed along to 50% of a carrier's offspring, on average.
Myotonic dystrophy is one of several known trinucleotide repeat disorders. Certain areas of DNA have repeated sequences of two or three nucleotides.
DM1
In DM1, the affected gene is called DMPK, which codes for myotonic dystrophy protein kinase,[5] a protein expressed predominantly in skeletal muscle.[6] The gene is located on the long arm of chromosome 19.[7]
In DM1, there is a repeat of the triplet cytosine-thymine-guanine (CTG) in the DMPK gene. The number of repeats varies greatly, but the average number in a healthy person is between 5 and 37. Additional trinucleotide repeats inserted during cell division can produce protein instability.[citation needed] Consequently, the repeat size of an individual with DM1 will become larger usually during gametogenesis or early embryonic development. This explains the phenomenon of anticipation, as each child of an affected adult will have a larger expansion than their parent. Individuals with larger expansions have an earlier onset of the disorder and a more severe phenotype.
DM2
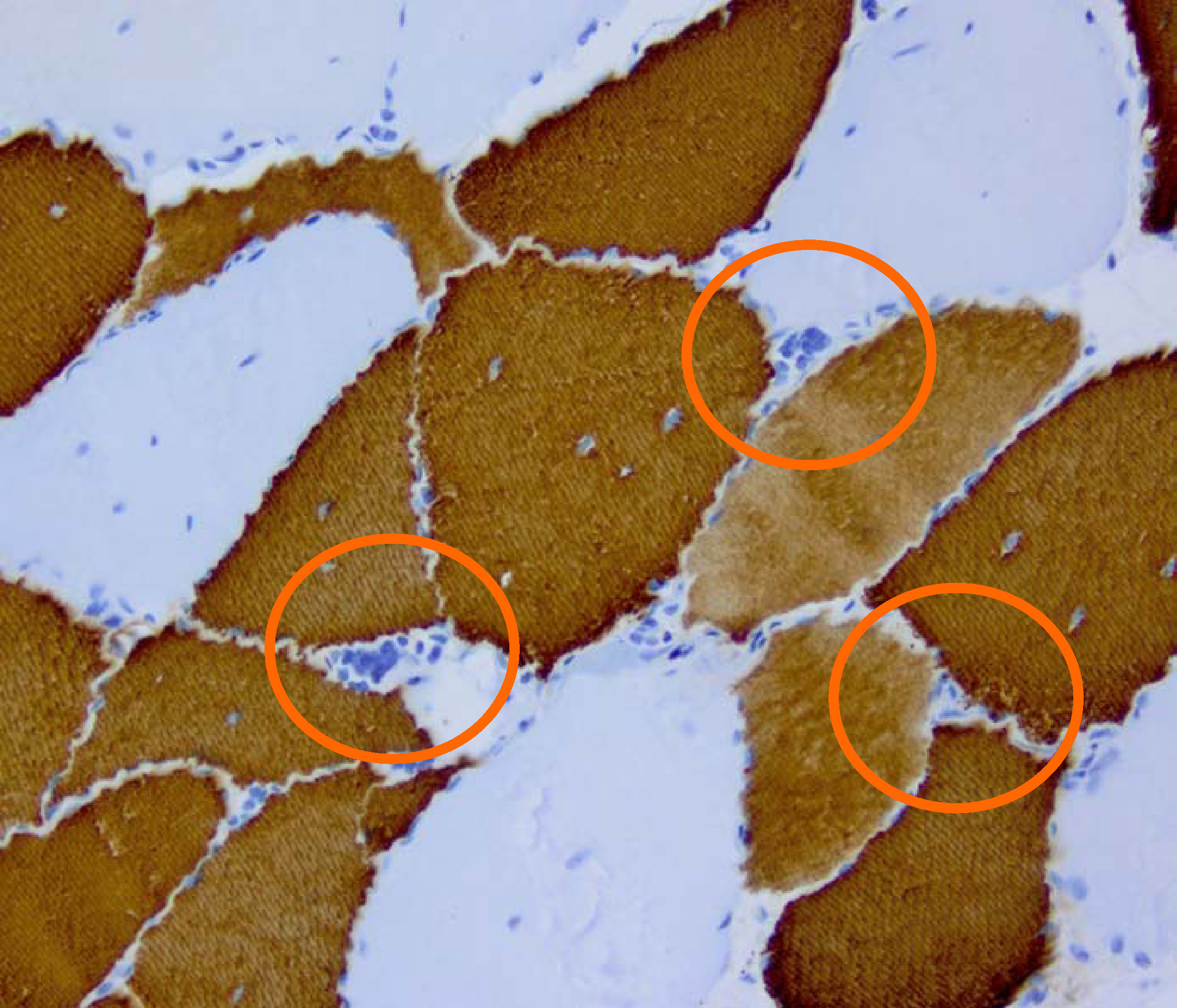
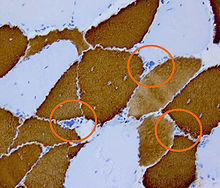 Histopathology of DM2. Muscle biopsy showing mild myopathic changes and grouping of atrophic fast fibres (type 2, highlighted). Immunohistochemical staining for type-1 ("slow") myosin
Histopathology of DM2. Muscle biopsy showing mild myopathic changes and grouping of atrophic fast fibres (type 2, highlighted). Immunohistochemical staining for type-1 ("slow") myosinDM2 is caused by a defect of the ZNF9 gene on chromosome 3.[8] The specific defect is a repeat of the cytosine-cytosine-thymine-guanosine (CCTG) tetranucleotide in the ZNF9 gene.[8] As it involves the repeat of four nucleotides, it is not a trinucleotide repeat disorder, but rather a tetranucleotide repeat disorder.[9]
The repeat expansion for DM2 is much larger than for DM1, ranging from 75 to over 11,000 repeats.[8] Unlike in DM1, the size of the repeated DNA expansion in DM2 does not appear to make a difference in the age of onset or disease severity.[citation needed] Anticipation appears to be less significant in DM2 and most current reviews only report mild anticipation as a feature of DM2.[citation needed]
Diagnosis
The diagnosis of DM1 and DM2 can be difficult due to the large number of neuromuscular disorders, most of which are very rare. More than 40 neuromuscular disorders exist with close to 100 variants.[citation needed]
As a result, patients with multiple symptoms that may be explained by a complex disorder such as DM1 or DM2 will generally be referred by their primary care physician to a neurologist for diagnosis. Depending on the presentation of symptoms, patients may be referred to a number of medical specialists including cardiologists, ophthalmologists, endocrinologists, and rheumatologists. In addition, the clinical presentation is obscured by the degree of severity or the presence of unusual phenotypes.
It is common that the clinical presentation for both DM1 and DM2 patients does not conform to the perceptions of these diseases held by many neurologists. Clinicians who are less familiar with the myotonic dystrophies in their day to day practice may expect patients with both forms to present with the more severe classic symptoms of DM1. As a result, patients may remain undiagnosed or be misdiagnosed.
Even though there is presently no cure for DM and management is currently symptom based, a precise diagnosis is still necessary because of multiple other problems that may develop over time, e. g. cataracts). An accurate diagnosis is important to assist with appropriate medical monitoring and medical management of symptoms. In addition, genetic counseling should be made available to all patients because of the high risk of transmission. Potentially serious anesthetic risks are important to note, so the presence of this disorder should be brought to the attention of all medical providers.
Management
There is currently no cure for or treatment specific to myotonic dystrophy. Complications of the disease, including heart problems, cataracts, and other abnormalities, can be treated but not cured. However there are medical interventions and medications that may relieve some of the symptoms such as myotonia, pain, and excessive sleepiness. Some treatments have been subject to systematic review for safety and efficacy through the Cochrane Reviews for symptoms such as hypersomnia (excessive daytime sleepiness), myotonia, strength training, aerobic exercise training, and foot drop.
Recent research has provided more information on the underlying molecular pathophysiologic mechanisms involved in myotonic dystrophy and has fueled interest and research into new approaches for more specific and effective treatment. Research in areas such as high-throughput screening and antisense therapy hold hope for more effective targeted treatments for the future. Altered splicing of the muscle-specific chloride channel 1 (ClC-1) has been shown to cause the myotonic phenotype of DM1 and is reversible in mouse models using Morpholino antisense to modify splicing of ClC-1 mRNA.[10]
Progress in this area is being fueled by the sharing of research by scientists and clinicians at biannual meetings by the International Myotonic Dystrophy Consortium (IDMC). The 7th biennial meeting IDMC-7 took place in Wurzburg, Germany September 9–12, 2009. The 8th biennial meeting will take place in Clearwater, Florida, USA November 30-December 3, 2011.
Screening
Screening for the repeat expansions on the DMPK gene for DM1 is targeted at chromosome 19, while the ZNF9 gene for DM2 is found on chromosome 3. Genetic tests, including prenatal testing, are available for both confirmed forms. Molecular testing is considered the gold standard of diagnosis. Further forms of myotonic dystrophy (DM3, DM4, and DMX) are suspected by researchers with possible defects on chromosome 16 and chromosome 21.[citation needed]
References
- ^ Le Ber I, Martinez M, Campion D, et al. (2004). "A non-DM1, non-DM2 multisystem myotonic disorder with frontotemporal dementia: phenotype and suggestive mapping of the DM3 locus to chromosome 15q21-24". Brain 127 (Pt 9): 1979–92. doi:10.1093/brain/awh216. PMID 15215218. http://brain.oxfordjournals.org/cgi/pmidlookup?view=long&pmid=15215218.
- ^ "Myotonic Dystrophy Type 2". http://www.ncbi.nlm.nih.gov/books/bv.fcgi?rid=gene.chapter.myotonic-d2. Retrieved 2008-02-24.
- ^ Udd B, Meola G, Krahe R, et al. (2006). "140th ENMC International Workshop: Myotonic Dystrophy DM2/PROMM and other myotonic dystrophies with guidelines on management". Neuromuscul. Disord. 16 (6): 403–13. doi:10.1016/j.nmd.2006.03.010. PMID 16684600. http://linkinghub.elsevier.com/retrieve/pii/S0960-8966(06)00097-6.
- ^ NIH Genetics Home Reference http://ghr.nlm.nih.gov/condition/myotonic-dystrophy
- ^ Mahadevan M, Tsilfidis C, Sabourin L, et al. (March 1992). "Myotonic dystrophy mutation: an unstable CTG repeat in the 3' untranslated region of the gene". Science 255 (5049): 1253–5. doi:10.1126/science.1546325. PMID 1546325. http://www.sciencemag.org/cgi/pmidlookup?view=long&pmid=1546325.
- ^ van der Ven PF, Jansen G, van Kuppevelt TH, et al. (November 1993). "Myotonic dystrophy kinase is a component of neuromuscular junctions". Human Molecular Genetics 2 (11): 1889–94. doi:10.1093/hmg/2.11.1889. PMID 8281152. http://hmg.oxfordjournals.org/cgi/pmidlookup?view=long&pmid=8281152.
- ^ Harley HG, Walsh KV, Rundle S, et al. (May 1991). "Localisation of the myotonic dystrophy locus to 19q13.2-19q13.3 and its relationship to twelve polymorphic loci on 19q". Human Genetics 87 (1): 73–80. doi:10.1007/BF01213096. PMID 2037285.
- ^ a b c Day JW, Ricker K, Jacobsen JF, et al. (February 2003). "Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum". Neurology 60 (4): 657–64. doi:10.1001/archneur.60.5.657. PMID 12601109. http://www.neurology.org/cgi/pmidlookup?view=long&pmid=12601109.
- ^ Liquori CL, Ricker K, Moseley ML, et al. (August 2001). "Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9". Science 293 (5531): 864–7. doi:10.1126/science.1062125. PMID 11486088. http://www.sciencemag.org/cgi/pmidlookup?view=long&pmid=11486088.
- ^ Wheeler TM, Lueck JD, Swanson MS, Dirksen RT, Thornton CA (2007). "Correction of ClC-1 splicing eliminates chloride channelopathy and myotonia in mouse models of myotonic dystrophy". J. Clin. Invest. 117 (12): 3952–7. doi:10.1172/JCI33355. PMC 2075481. PMID 18008009. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2075481.
External links
- Myotonic dystrophy at NLM Genetics Home Reference
- GeneReview/NCBI/NIH/UW entry on Myotonic Dystrophy Type 1
- GeneReview/NCBI/NIH/UW entry on Myotonic Dystrophy Type 2
- Searchable database at Dutch Neuromuscular Research
- 140th ENMC International Workshop 2006 Myotonic Dystrophy DM2/PROMM and other Myotonic dystrophies]
- Information from the Myotonic Dystrophy Foundation
- Information from the International Myotonic Dystrophy Organization (IMDO)
- MDSG Information
- Information from the Neuromuscular Disease Center
- DM Toolbox Research tools for Myotonic Dystrophy from the Marigold Foundation
Muscular dystrophy The Nine Primary Muscular Dystrophies Congenital • dystrophin (Becker's, Duchenne) • Distal • Emery-Dreifuss • Facioscapulohumeral • Limb-girdle muscular dystrophy • Myotonic • OculopharyngealRelated topicsNational/International Organizations US government Institutes and Legislation National/International Events Recent or Ongoing Clinical Trials Diseases of myoneural junction and muscle / neuromuscular disease (G70–G73, 358–359) Neuromuscular-
junction diseaseMyopathy/
congenital myopathyADAROther structuralOtherOtherNon-Mendelian inheritance: anticipation Trinucleotide Polyglutamine (PolyQ), CAGNon-PolyglutamineCGG (Fragile X syndrome) · GAA (Friedreich's ataxia) · CTG (Myotonic dystrophy type 1) · CTG (Spinocerebellar ataxia 8) · CAG (Spinocerebellar ataxia 12)Tetranucleotide CCTG (Myotonic dystrophy type 2)Pentanucleotide ATTCT (Spinocerebellar ataxia 10)Deficiencies of intracellular signaling peptides and proteins GTP-binding protein regulators GTPase-activating proteinMarinesco–Sjögren syndrome · Aarskog–Scott syndrome · Juvenile primary lateral sclerosis · X-Linked mental retardation 1G protein cAMP/GNAS1: Pseudopseudohypoparathyroidism · Progressive osseous heteroplasia · Pseudohypoparathyroidism · Albright's hereditary osteodystrophy · McCune–Albright syndrome
CGL 2RAS: HRAS (Costello syndrome) · KRAS (Noonan syndrome 3, KRAS Cardiofaciocutaneous syndrome)
RAB: RAB7 (Charcot–Marie–Tooth disease) · RAB23 (Carpenter syndrome) · RAB27 (Griscelli syndrome type 2)
RHO: RAC2 (Neutrophil immunodeficiency syndrome)
ARF: SAR1B (Chylomicron retention disease) ARL13B (Joubert syndrome 8) · ARL6 (Bardet–Biedl syndrome 3)MAP kinase Other kinase/phosphatase RPS6KA3 (Coffin-Lowry syndrome) · CHEK2 (Li-Fraumeni syndrome 2) · IKBKG (Incontinentia pigmenti) · STK11 (Peutz–Jeghers syndrome) · DMPK (Myotonic dystrophy 1) · ATR (Seckel syndrome 1) · GRK1 (Oguchi disease 2) · WNK4/WNK1 (Pseudohypoaldosteronism 2)PTEN (Bannayan–Riley–Ruvalcaba syndrome, Lhermitte–Duclos disease, Cowden syndrome, Proteus-like syndrome) · MTM1 (X-linked myotubular myopathy) · PTPN11 (Noonan syndrome 1, LEOPARD syndrome, Metachondromatosis)Signal transducing adaptor proteins Other NF2 (Neurofibromatosis type II) · NOTCH3 (CADASIL) · PRKAR1A (Carney complex) · PRKAG2 (Wolff–Parkinson–White syndrome) · PRKCSH (PRKCSH Polycystic liver disease) · XIAP (XIAP2)see also intracellular signaling peptides and proteins
B structural (perx, skel, cili, mito, nucl, sclr) · DNA/RNA/protein synthesis (drep, trfc, tscr, tltn) · membrane (icha, slcr, atpa, abct, othr) · transduction (iter, csrc, itra), trfkGenetic disorder, protein biosynthesis: Transcription factor/coregulator deficiencies (1) Basic domains (2) Zinc finger
DNA-binding domains2.1 (Intracellular receptor): Thyroid hormone resistance · Androgen insensitivity syndrome (PAIS, MAIS, CAIS) · Kennedy's disease · PHA1AD pseudohypoaldosteronism · Estrogen insensitivity syndrome · X-linked adrenal hypoplasia congenita · MODY 1 · Familial partial lipodystrophy 3 · SF1 XY gonadal dysgenesis
2.2: Barakat syndrome · Tricho–rhino–phalangeal syndrome
2.3: Greig cephalopolysyndactyly syndrome/Pallister-Hall syndrome · Denys–Drash syndrome · Duane-radial ray syndrome · MODY 7 · MRX 89 · Townes–Brocks syndrome · Acrocallosal syndrome · Myotonic dystrophy 2
2.5: Autoimmune polyendocrine syndrome type 1(3) Helix-turn-helix domains 3.1: ARX (Ohtahara syndrome, Lissencephaly X2) · HLXB9 (Currarino syndrome) · HOXD13 (SPD1 Synpolydactyly) · IPF1 (MODY 4) · LMX1B (Nail–patella syndrome) · MSX1 (Tooth and nail syndrome, OFC5) · PITX2 (Axenfeld syndrome 1) · POU4F3 (DFNA15) · POU3F4 (DFNX2) · ZEB1 (Posterior polymorphous corneal dystrophy 3, Fuchs' dystrophy 3) · ZEB2 (Mowat-Wilson syndrome)
3.2: PAX2 (Papillorenal syndrome) · PAX3 (Waardenburg syndrome 1&3) · PAX4 (MODY 9) · PAX6 (Gillespie syndrome, Coloboma of optic nerve) · PAX8 (Congenital hypothyroidism 2) · PAX9 (STHAG3)
3.3: FOXC1 (Axenfeld syndrome 3, Iridogoniodysgenesis, dominant type) · FOXC2 (Lymphedema–distichiasis syndrome) · FOXE1 (Bamforth–Lazarus syndrome) · FOXE3 (Anterior segment mesenchymal dysgenesis) · FOXF1 (ACD/MPV) · FOXI1 (Enlarged vestibular aqueduct) · FOXL2 (Premature ovarian failure 3) · FOXP3 (IPEX)
3.5: IRF6 (Van der Woude syndrome, Popliteal pterygium syndrome)(4) β-Scaffold factors
with minor groove contacts4.2: Hyperimmunoglobulin E syndrome
4.3: Holt-Oram syndrome · Li-Fraumeni syndrome · Ulnar–mammary syndrome
4.7: Campomelic dysplasia · MODY 3 · MODY 5 · SF1 (SRY XY gonadal dysgenesis, Premature ovarian failure 7) · SOX10 (Waardenburg syndrome 4c, Yemenite deaf-blind hypopigmentation syndrome)
4.11: Cleidocranial dysostosis(0) Other transcription factors Ungrouped Transcription coregulators see also transcription factors
B structural (perx, skel, cili, mito, nucl, sclr) · DNA/RNA/protein synthesis (drep, trfc, tscr, tltn) · membrane (icha, slcr, atpa, abct, othr) · transduction (iter, csrc, itra), trfkCategories:- Autosomal dominant disorders
- Muscular dystrophy
- Myoneural junction and neuromuscular diseases
Wikimedia Foundation. 2010.