- Duchenne muscular dystrophy
-
Duchenne muscular dystrophy Classification and external resources 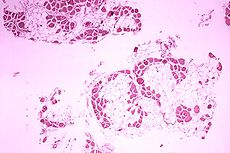
Histopathology of gastrocnemius muscle from patient who died of pseudohypertrophic muscular dystrophy, Duchenne type. Cross section of muscle shows extensive replacement of muscle fibers by adipose cells.ICD-10 G71.0 ICD-9 359.1 OMIM 310200 DiseasesDB 3985 MedlinePlus 000705 MeSH D020388 Duchenne muscular dystrophy (DMD) is a recessive X-linked form of muscular dystrophy, which results in muscle degeneration, difficulty walking, breathing, and death. The incidence is 1 in 3,000 boys.[1] Females and males are affected, though females are rarely affected and are more often carriers. The disorder is caused by a mutation in the dystrophin gene, located in humans on the X chromosome (Xp21). The dystrophin gene codes for the protein dystrophin, an important structural component within muscle tissue. Dystrophin provides structural stability to the dystroglycan complex (DGC), located on the cell membrane.
Symptoms usually appear in male children before age 5 and may be visible in early infancy. Progressive proximal muscle weakness of the legs and pelvis associated with a loss of muscle mass is observed first. Eventually this weakness spreads to the arms, neck, and other areas. Early signs may include pseudohypertrophy (enlargement of calf and deltoid muscles), low endurance, and difficulties in standing unaided or inability to ascend staircases. As the condition progresses, muscle tissue experiences wasting and is eventually replaced by fat and fibrotic tissue (fibrosis). By age 10, braces may be required to aid in walking but most patients are wheelchair dependent by age 12. Later symptoms may include abnormal bone development that lead to skeletal deformities, including curvature of the spine. Due to progressive deterioration of muscle, loss of movement occurs, eventually leading to paralysis. Intellectual impairment may or may not be present but if present, does not progressively worsen as the child ages. The average life expectancy for patients afflicted with DMD varies from late teens to early to mid 20s. There have been reports of a few DMD patients surviving to the age of 40, but this is extremely rare.[citation needed]
Contents
Incidence/prevalence
Duchenne muscular dystrophy is caused by mutations in the dystrophin gene, which is located on the X chromosome. Due to this, DMD has an incidence of 1 in 4,000[2] newborn males. Mutations within the dystrophin gene can either be inherited or occur spontaneously during germline transmission.
Eponym
DMD is named after the French neurologist Guillaume Benjamin Amand Duchenne (1806–1875), who first described the disease in 1861[3] though it had already been described by the Neapolitan physicians Giovanni Semmola in 1834 and Gaetano Conte in 1836[4][5].
Pathogenesis
Duchenne muscular dystrophy is caused by a mutation of the dystrophin gene at locus Xp21. Dystrophin is responsible for connecting the cytoskeleton of each muscle fiber to the underlying basal lamina (extracellular matrix) through a protein complex containing many subunits. The absence of dystrophin permits excess calcium to penetrate the sarcolemma (cell membrane).[6] Alterations in these signalling pathways cause water to enter into the mitochondria which then burst. In skeletal muscle dystrophy, mitochondrial dysfunction gives rise to an amplification of stress-induced cytosolic calcium signals and an amplification of stress-induced reactive-oxygen species (ROS) production. In a complex cascading process that involves several pathways and is not clearly understood, increased oxidative stress within the cell damages the sarcolemma and eventually results in the death of the cell. Muscle fibers undergo necrosis and are ultimately replaced with adipose and connective tissue.
Symptoms
The main symptom of Duchenne muscular dystrophy, a progressive neuromuscular disorder, is muscle weakness associated with muscle wasting with the voluntary muscles[citation needed] being first affected, especially affecting the muscles of the hips, pelvic area, thighs, shoulders, and calf muscles. Muscle weakness also occurs in the arms, neck, and other areas, but not as early as in the lower half of the body. Calves are often enlarged. Symptoms usually appear before age 6 and may appear as early as infancy. The other physical symptoms are:
- Awkward manner of walking, stepping, or running. (patients tend to walk on their forefeet, because of an increased calf tonus. Also, toe walking is a compensatory adaptation to knee extensor weakness.)
- Frequent falls
- Fatigue
- Difficulty with motor skills (running, hopping, jumping)
- Increased Lumbar lordosis, leading to shortening of the hip-flexor muscles. This has an effect on overall posture and a manner of walking, stepping, or running.
- Muscle contractures of achilles tendon and hamstrings impair functionality because the muscle fibers shorten and fibrosis occurs in connective tissue
- Progressive difficulty walking
- Muscle fiber deformities
- Pseudohypertrophy (enlarging) of tongue and calf muscles. The muscle tissue is eventually replaced by fat and connective tissue, hence the term pseudohypertrophy.
- Higher risk of neurobehavioral disorders (e.g., ADHD, Autistic-Spectrum Disorders), learning disorders (dyslexia), and non-progressive weaknesses in specific cognitive skills (in particular short-term verbal memory), which are believed to be the result of absent or dysfunctional dystrophin in the brain.
- Eventual loss of ability to walk (usually by the age of 12)
- Skeletal deformities (including scoliosis in some cases)
Signs and tests
Muscle wasting begins in the legs and pelvis, then progresses to the muscles of the shoulders and neck, followed by loss of arm muscles and respiratory muscles. Calf muscle enlargement (pseudohypertrophy) is quite obvious. Cardiomyopathy(DCM) is common, but the development of congestive heart failure or arrhythmias (irregular heartbeats) is only occasional.
- A positive Gowers' sign reflects the more severe impairment of the lower extremities muscles. The child helps himself to get up with upper extremities: first by rising to stand on his arms and knees, and then "walking" his hands up his legs to stand upright.
- Affected children usually tire more easily and have less overall strength than their peers.
- Creatine kinase (CPK-MM) levels in the bloodstream are extremely high.
- An electromyography (EMG) shows that weakness is caused by destruction of muscle tissue rather than by damage to nerves.
- Genetic testing can reveal genetic errors in the Xp21 gene.
- A muscle biopsy (immunohistochemistry or immunoblotting) or genetic test (blood test) confirms the absence of dystrophin, although improvements in genetic testing often make this unnecessary.
Diagnosis
DNA test
The muscle-specific isoform of the dystrophin gene is composed of 79 exons, and DNA testing and analysis can usually identify the specific type of mutation of the exon or exons that are affected. DNA testing confirms the diagnosis in most cases.[7]
Muscle biopsy
If DNA testing fails to find the mutation, a muscle biopsy test may be performed. A small sample of muscle tissue is extracted (usually with a scalpel instead of a needle) and a dye is applied that reveals the presence of dystrophin. Complete absence of the protein indicates the condition.
Over the past several years DNA tests have been developed that detect more of the many mutations that cause the condition, and muscle biopsy is not required as often to confirm the presence of Duchenne's.
Prenatal tests
DMD is carried by an X-linked recessive gene. Males have only one X chromosome, so one copy of the mutated gene will cause DMD. Fathers cannot pass X-linked traits on to their sons, so the mutation is transmitted by the mother.[8]
If the mother is a carrier, and therefore one of her two X chromosomes has a DMD mutation, there is a 50% chance that a female child will inherit that mutation as one of her two X chromosomes, and be a carrier. There is a 50% chance that a male child will inherit that mutation as his one X chromosome, and therefore have DMD.
Prenatal tests can tell whether their unborn child has the most common mutations, and they may choose to have an abortion. There are many mutations responsible for DMD, and some have not been identified, so genetic testing only works when family members with DMD have a mutation that has been identified.
Chorion villus sampling (CVS) can be done at 11–14 weeks, and has a 2% risk of miscarriage. Amniocentesis can be done after 15 weeks, and has a 0.5% risk of miscarriage. Fetal blood sampling can be done at about 18 weeks.[citation needed] Another option in the case of unclear genetic test results is fetal muscle biopsy.
Treatment
There is no current cure for DMD, although phase 1-2a trials with exon-skipping treatment for certain mutations have halted decline and produced small clinical improvements in walking.
Treatment is generally aimed at controlling the onset of symptoms to maximize the quality of life, and include the following:
- Corticosteroids such as prednisolone and deflazacort increase energy and strength and defer severity of some symptoms.
- Randomised control trials have shown that beta2-agonists increase muscle strength but do not modify disease progression. Follow-up time for most RCTs on beta2-agonists is only around 12 months and hence results cannot be extrapolated beyond that time frame.[citation needed]
- Mild, non-jarring physical activity such as swimming is encouraged. Inactivity (such as bed rest) can worsen the muscle disease.
- Physical therapy is helpful to maintain muscle strength, flexibility, and function.
- Orthopedic appliances (such as braces and wheelchairs) may improve mobility and the ability for self-care. Form-fitting removable leg braces that hold the ankle in place during sleep can defer the onset of contractures.
- Appropriate respiratory support as the disease progresses is important
Comprehensive multi-disciplinary care standards/guidelines for DMD have been developed by the Centers for Disease Control and Prevention (CDC), and were published in two parts in The Lancet Neurology in 2010. To download the two articles in PDF format, go to the TREAT-NMD website: http://www.treat-nmd.eu/patients/DMD/dmd-care/
Prognosis
Duchenne muscular dystrophy eventually affects all voluntary muscles and involves the heart and breathing muscles in later stages. The life expectancy typically ranges from the late teens to the mid-20s.[9] Recent advancements in medicine are extending the lives of those afflicted.
In rare cases, persons with DMD have been seen to survive into the forties or early fifties, with the use of proper positioning in wheelchairs and beds, ventilator support (via tracheostomy or mouthpiece), airway clearance, and heart medications, if required.[citation needed] Early planning of the required supports for later-life care has shown greater longevity in people living with DMD.
Physical therapy
Physical therapists are concerned with enabling children to reach their maximum physical potential. Their aim is to:
- minimize the development of contractures and deformity by developing a programme of stretches and exercises where appropriate
- anticipate and minimize other secondary complications of a physical nature
- monitor respiratory function and advise on techniques to assist with breathing exercises and methods of clearing secretions
- Schedule weekly to monthly sessions at a massage therapist to alleviate pain present.
Mechanical ventilatory/respiration assistance
Modern "volume ventilators/respirators," which deliver an adjustable volume (amount) of air to the person with each breath, are valuable in the treatment of people with muscular dystrophy related respiratory problems. The ventilator may require an invasive endotracheal or tracheotomy tube through which air is directly delivered, but, for some people non-invasive delivery through a face mask or mouthpiece is sufficient. Positive airway pressure machines, particularly bi-level ones, are sometimes used in this latter way. The respiratory equipment may easily fit on a ventilator tray on the bottom or back of a power wheelchair with an external battery for portability.
Ventilator treatment may start in the mid to late teens when the respiratory muscles can begin to collapse. If the vital capacity has dropped below 40 percent of normal, a volume ventilator/respirator may be used during sleeping hours, a time when the person is most likely to be under ventilating ("hypoventilating"). Hypoventilation during sleep is determined by a thorough history of sleep disorder with an oximetry study and a capillary blood gas (See Pulmonary Function Testing).
If the vital capacity continues to decline to less than 30 percent of normal, a volume ventilator/respirator may also be needed during the day for more assistance. The person gradually will increase the amount of time using the ventilator/respirator during the day as needed.
However, there are also people with the disease in their 20's who have no need for a ventilator.
Occupational and Physical Therapy
The goal of occupational therapy is to obtain a clear understanding of the individual, of their social circumstances, and of their environment in order to develop a treatment plan that will improve their quality of life.[10] Individuals with DMD often experience difficulties in areas of self-care, productivity, and leisure. This is related to the effects of the disorder, such as decreased mobility, decreased strength and postural stability, progressive deterioration of upper-limb function, and contractures.[10] Occupational and physical therapists address an individual's limitations using meaningful occupations and by grading the activity, by using different assessments and resources such as splinting, bracing, manual muscle testing (MMT), ROM, postural intervention, and equipment prescription.[11]
Splinting and Orthoses
Splints, also referred to as orthoses, are designed to maintain or improve ROM, prevent deformity, and improve function. Splints help to support and keep limbs stretched, which delays or prevents the onset of contractures that commonly affect the knees, hips, feet, elbows, wrists, and fingers.[11] An ankle foot orthoses (AFO) can be used during sleep or during the day. The purpose of this is to keep the foot from pointing downward and sustain the stretch of the achilles tendon.[11] Maintaining the length of the tendo-achilles, also referred to as the gastrocnemius-soleus complex, which is extremely important for walking. Knee ankle foot orthoses (KAFO) are also used for walking or for standing and can be used to prolong ambulation or help delay the onset of lower limb contractures.[11]
Manual Muscle Testing and Range of Motion
MMT is used to evaluate muscular strength, whereas goniometry or ROM tests measure movement around a joint. These tests indicate need for intervention such as passive and active ROM, strengthening and splinting. Passive ROM combined with the use of night splints can significantly improve tendo-achilles contractures.[12]
Seating and Positioning
Proper seating is essential to prevent spinal curvatures. Severe scoliosis is common in DMD and can interfere with sitting, sleeping, and breathing.[13] A wheelchair that is fitted appropriately accounts for frame size, type of seat, lumbar support, and cushioning to avoid pressure ulcers.[10] It should be equipped with other mechanical devices, such as tilt ability, in order to provide comfort and to protect the skin. Power wheelchairs are indicated for most clients who can no longer ambulate, as they do not have enough upper extremity strength to propel a manual wheelchair independently.[10] DMD affects many people in their adolescence, therefore, it is crucial for occupational therapists to be conscious that significant development may occur during this time.[13] Without proper seating and postural support throughout development, deformation may occur, which could result in dysfunctional positioning. It is important for occupational therapists to re-evaluate the fit of an individual’s wheelchair as often as every year during adolescence <.[13]
Adaptive Equipment and Devices
There are many alternate mobility options, positioning aids, and other equipment that occupational therapists may prescribe. These include walkers or quad-canes, which can be used for individuals who are able ambulate to reduce the risk of falling. In addition, transfer boards, mechanical lifts, and specific transferring education are important because fractures have been seen to occur during transfers as a result of osteoporosis.[14] Handheld shower heads and bath benches are indicated to enable individuals to manage their own self-care needs as much as possible.[14] Individuals who are able to bear weight and take a few steps may utilize commode chairs, thus giving them the ability to toilet independently.[14] To complement an individualized wheelchair, an occupational therapist may also consider prescribing a hospital bed, pressure-relieving mattresses, and foam wedges for proper positioning to prevent pressure skin ulcers, contractures, and deformities.[10] Specialized trays, input devices and software may be prescribed to facilitate computer use as well.
Social Skills Development
Along with physical difficulties, individuals with DMD may have social issues that an occupational therapist can assist them in overcoming.[14] Group sessions or individualized programs that focus on coping mechanisms for depression are examples of what an occupational therapist can facilitate.[14] Self-esteem building for individuals of all ages is an essential part of ensuring that a high quality of life is achieved.[14] Occupational therapy intervention can play an essential role in supporting the development of social skills through group interactions and other life experiences.[14] An occupational therapist can use a variety of psychosocial frameworks for developing strategies and techniques for individuals to utilize, which will help them work through various psychosocial issues they may be experiencing.[10]
Sexual Health
Sexuality is a topic that many people feel uncomfortable discussing and thus may be overlooked by health care professionals. An occupational therapist will educate individuals with DMD on safe and effective ways to experience their sexual life.[10] Such education can include various forms of sex as well as numerous positions that they would be able to perform.[10]
Employment
Gaining and maintaining employment can be difficult for individuals with DMD. An occupational therapist may collaborate with an individual, employer, and case manager to ensure that the individual’s work environment is as enabling and accessible as possible.[10] By adapting the physical work environment, the social environment, and the work requirements and guidelines, an individual can maintain meaningful employment as well as be as an asset to his or her employer. This may not only impact the individual’s perceived self-efficacy but also his or her financial well being.[10]
Home Modifications
Maintaining independence is often a main focus of occupational therapy interventions if it is a priority of the client. Within the home, there are numerous obstacles that may prevent a client from being as independent as possible. Home modifications and adaptations are something that an occupational therapist can assist with.[10] Such modifications may include: railings for safe mobility and transfers, lifts, adapted kitchens that are accessible for wheelchairs, and bathroom modifications such as raised toilet seats or modified baths.[10] For example providing adaptive equipment for playing computer and video games, supports for biking and adaptations for fishing rods.
Leisure
An occupational therapist can support individuals with DMD to find leisure activities that are meaningful for them to participate in; accommodations and adaptations can be made to enhance participation.[10]
Advocacy
In order to create a successful therapeutic relationship it is important to coordinate with family members, friends, and other social resources to ensure that a person with DMD has both physical and emotional support. An occupational therapist can act as an advocate for his or her client and can educate those around him or her regarding information about the illness, supports, resources, and other concerns.[10] An occupational therapist can also provide his or her client with the tools to learn how to advocate for him or herself.[10]
Ongoing research
Current research includes exon-skipping, stem cell replacement therapy, analog up-regulation, gene replacement and supportive care to slow disease progression.
Exon-skipping
Antisense oligonucleotides, structural analogs of DNA, are the basis of a potential therapy for patients afflicted with DMD. Two kinds of antisense oligos, Morpholino oligos and 2'-O-methyl phosphorothioate oligos, have been tested in early-phase clinical trials for DMD and have restored some dystrophin expression in muscles of DMD patients with a particular class of DMD-causing mutations. Further clinical trials are proceeding with these compounds.
Oligo-mediated exon skipping has resulted in clinical improvement in 12 patients in a Phase 1-2a study. On a standard test, the 6-minute walk test, patients whose performance had been declining instead improved, from 385 meters to 420 meters.[15][16] DMD may result from mRNA that contains out-of-frame mutations (e.g. deletions, insertions or splice site mutations), resulting in frameshift or early termination so that in most muscle fibers no functional dystrophin is produced (though some revertant muscle fibers produce some dystrophin). In many cases an antisense oligonucleotide can be used to trigger skipping of an adjacent exon to restore the reading frame and production of partially-functional dystrophin.
Patients with Becker's muscular dystrophy, which is milder than DMD, have a form of dystrophin which is functional even though it is shorter than normal dystrophin.[17] In 1990 England et al. noticed that a patient with mild Becker muscular dystrophy was lacking 46% of his coding region for dystrophin.[17] This functional, yet truncated, form of dystrophin gave rise to the notion that shorter dystrophin can still be therapeutically beneficial. Concurrently, Kole et al. had modified splicing by targeting pre-mRNA with antisense oligonucleotides (AONs).[18] Kole demonstrated success using splice-targeted AONs to correct missplicing in cells removed from beta-thalassemia patients[19][20] Wilton's group tested exon skipping for muscular dystrophy.[21][22] Successful preclinical research led to the current efforts to use splice-modifying oligos to change DMD dystrophin to a more functional form of dystrophin, in effect converting Duchenne MD into Becker MD.
Though AONs hold promise, one of their major pitfalls is the need for periodic redelivery into muscles. Systemic delivery on a recurring basis is being tested in humans (http://clinicaltrials.gov/ct2/show/NCT00844597). To circumvent the requirement for periodic oligo delivery, a long-term exon-skip therapy is being explored. This therapy consists of modifying the U7 small nuclear RNA at the 5' end of the non-translated RNA to target regions within pre-mRNA. This has been shown to work in the DMD equivalent mouse, mdx.[23]
Stem cell replacement
Though stem cells isolated from the muscle (satellite cells) have the ability to differentiate into myotubes when injected directly into the muscle of animals, they lack the ability to spread systemically throughout. To effectively deliver a therapeutic dose to an isolated muscle it would require direct injections to that muscle every 2mm.[24] This problem was circumvented by using another multipotent stem cell, termed pericytes, that are located within the blood vessels of skeletal muscle. These cells have the ability to be delivered systemically and uptaken by crossing the vascular barrier. Once past the vasculature, pericytes have the ability to fuse and form myotubes.[25] This means that they can be injected arterially, crossing through arterial walls into muscle, where they can differentiate into potentially functional muscle. These findings show potential for stem cell therapy of DMD. The pericyte-derived cells would be extracted, grown in culture, and then these cells would be injected into the blood stream where the possibility exists that they might find their way into injured regions of skeletal muscle.
Utrophin upregulation
Upregulation of utrophin, whose gene is located on chromosome 6, can partially compensate for the lack of dystrophin in mice. Human testing is planned.[citation needed].
Supportive care - drug development
Recent research shows Losartan, a currently available drug used for treating hypertension, to be effective in halting the progress of the disease in mice that were genetically engineered to have Duchenne's.[26] Human trials are in planning.
Research from a group in France led by L. Ségalat has identified a number of drugs that are currently licensed for other applications as halting or reducing dramatically the advance of muscle degeneration in a worm model of DMD.[27] They are now using mouse models to confirm these findings, which so far are looking very promising, confirming the efficacy of these drugs. However, work in mice seems to be moving slowly. The main classes of drugs they identified were SSRI (i.e. antidepressants such as Prozac) and muscle relaxants, such as those used by athletes after heavy training. There is conflicting evidence from animal models suggesting that doing less exercise slows down the rate of degeneration of the muscle; therefore there is a possibility that both these drugs act somewhat as sedatives, although the reality seems to be that the worms and mice are more active overall, as they have less muscle damage and so can remain active for much longer.
More recently, a group at the Montreal Heart Institute and McGill University reported that a mouse model of Duchenne's muscular dystrophy demonstrated early metabolic alterations that precede overt cardiomyopathy and may represent an early "subclinical" signature of a defective nitric oxide (NO)/cGMP pathway. Accordingly, they used genetic and pharmacological approaches to test the hypothesis that enhancing cGMP, downstream of NO formation, improves the contractile function, energy metabolism, and sarcolemmal integrity. Treatment with Sildenafil delayed the appearance of symptoms in mouse hearts with Duchenne's and allowed to withstand an acutely increased cardiac workload.[28]
Gene therapy
In 2007, a team of researchers led by Jerry Mendell, M.D., at Nationwide Children's Hospital did the world's first clinical (viral-mediated) gene therapy trial for Duchenne MD.[29]
Scientific research published on 15 April 2010 from the Université Laval's Faculty of Medicine and the CHUQ Research Center indicates it is possible to repair the gene associated with causing DMD through the use of meganuclease enzymes though significant work remains until it can be tested in human patients.[30]
Biostrophin is a delivery vector for gene therapy in the treatment of Duchenne muscular dystrophy and Becker muscular dystrophy.[31]
Clinical trials
While PTC124 showed promising results in mice,[32][33] the Phase II trial was suspended when participants did not show significant increases in the six minute walk distance.[34]
Safety and efficacy studies of antisense oligonucleotides for exon skipping in Duchenne muscular dystrophy with Morpholino oligos[35] and with 2'-O-methyl phosphorothioate oligos[36] are in progress.
ACE-031 is in phase IIa. Patients with DMD are currently being enrolled to continue the clinical trail that is being sponsored by Acceleron Pharma, Inc with the financial help of Shire who paid $45 million upfront to enroll candidates. The estimated study completion date is set to be February 2012.[37][38]
Currently, this is the most promising successful treatment for DMD and similar disorders.
Counseling
Genetic counseling is advised for people with a family history of the disorder. Duchenne muscular dystrophy can be detected with about 95% accuracy by genetic studies performed during pregnancy.
Organizations specific to DMD
In addition to charities devoted to muscular dystrophies in general (such as MDA), these charities are devoted exclusively to DMD:
- Jesse's Journey - The Foundation for Gene and Cell Therapy: Jesse's Journey is located in London, Ontario, Canada working with families across North America and funding Duchenne research projects worldwide.
- The Foundation to Eradicate Duchenne: The Foundation to Eradicate Duchenne is a parent foundation near Washington, DC dedicated to funding research to help this generation of Duchenne Muscular Dystrophy boys.
- United Parent Project Muscular Dystrophy: United Parent Projects MD is an international organisation that has been set up by parents and friends of boys with DMD.
- Parent Project Muscular Dystrophy: Parent Project Muscular Dystrophy’s mission is to improve the treatment, quality of life and long-term outlook for all individuals affected by Duchenne muscular dystrophy (DMD) through research, advocacy, education and compassion.
- Charley's Fund: an organisation whose mission is to fund research for cure or treatment for Duchenne. Charley's Fund invests money in translational research – research that focuses on moving science from the lab into human clinical trials.
- Darius Goes West Foundation: a foundation revolving around the documentary film titled Darius Goes West (DGW). DGW won over 25 film festival awards in 2007, making it the most honored film on the circuit that year. It features the story of Darius Weems, a fifteen-year-old freshman with DMD, who leaves home with his eleven best friends in an attempt to see his wheelchair customized on MTV's Pimp my ride. Watch Trailer. Every DVD sold raises $17 for DMD research.
- Jett Foundation for Duchenne muscular dystrophy: New England based organization dedicated to raising funds and awareness for DMD and providing families with resources, like access to adaptive sports programs, that help boys with DMD to lead full and healthy lives.
- CureDuchenne: is a non-profit organization that aggressively funds leading edge research for treatments and a cure for Duchenne muscular dystrophy. Green Bay Packer Pro Bowl linebacker Clay Matthews has supported this organization in a television commercial.
- Action Duchenne: exclusively funds research for a cure and promotes campaigns for better medical care for Duchenne and Becker Muscular Dystrophy.
- DREAM Foundation: a Louisville, KY based organization devoted to raising funds and awareness. In addition, the foundation funded (through donation) the construction of five playgrounds in the Louisville area specifically designed for use by children with DMD.
- DuchenneConnect: A web-based registry for individuals with Duchenne/Becker, care providers, and members of the DBMD community. The website provides resources to assist with early, appropriate and least invasive diagnosis; to understand the benefits and limitations of genetic testing; to assist in the understanding and development of new treatment trials for Duchenne/Becker muscular dystrophy; and access to information and services for care, treatment, and genetic counseling.
- Save Our Sons : A Sydney, Australia based NON-profit organisation that holds many events to raise funds and awareness of DMD.
- The Shakti Foundation : The Shakti Foundation identified with the Adhiparasakthi Medical and Research Hospital as its sole project. This hospital is situated 92 km from Chennai, India and caters to the rural poor. The treatment is totally free. The Foundation has equipped this hospital in totality.
Famous sufferers of DMD
Alfredo Ferrari (born January, 1932 in Modena), nicknamed Alfredino or Dino, was the son of Enzo Ferrari. He designed the 1.5 L DOHC V6 engine for F2 at the end of 1955. Dino would never see the engine; he died 30 June 1956 in Modena at the age of 24, before his namesake automobiles Fiat Dino and Dino (automobile) were produced.
References
- ^ Duchenne and Becker Muscular Dystrophies, Merck Manual, Home Edition
- ^ Louise V. B. Anderson; Katharine M. D. Bushby (2001). of DMD Muscular Dystrophy: Methods and Protocols (Methods in Molecular Medicine). Totowa, NJ: Humana Press. p. 111. ISBN 0-89603-695-2. http://books.google.com/books?id=wR3g5u6Jwo4C&printsec=frontcover&dq=isbn:0896036952&cd=1#v=snippet&q=incidence of DMD.
- ^ doctor/950 at Who Named It?
- ^ http://www.cardiomiologia.unina2.it/index.php?option=com_content&view=article&id=54&Itemid=58
- ^ http://www.uildm.org/archivio_dm/156/scienza/37conteweb.shtml
- ^ http://www.forschungsportal.ch/unibe/abstracts/A_62193673.html
- ^ University of Utah Muscular Dystrophy
- ^ Duchenne and Becker muscular dystrophy, National Institutes of health
- ^ Duchenne Muscular Dystrophy (DMD)
- ^ a b c d e f g h i j k l m n o Stone, K; Tester, C; Howarth, A; Johnston, R; Traynor, N; McAndrew (2007). Occupational therapy and Duchenne muscular dystrophy. New Jersey: John Wiley & Sons Ltd.
- ^ a b c d Kinali, M; Manzur, AY; Muntoni, F (2008). "Recent developments in the management of Duchenne muscular dystrophy". Paediatrics and Child Health 18 (1): 22–26. doi:10.1016/j.paed.2007.10.001
- ^ Hyde, SA; et al. (2000). A randomized comparative study of two methods for controlling Tendo Achilles contracture in Duchenne muscular dystrophy. 10. pp. 257–263
- ^ a b c Richardson, M; Frank, A (2009). "Electric powered wheelchairs for those with muscular dystrophy: Problems of posture, pain and deformity". Disability and Rehabilitation: Assistive Technology 4 (3): 181–188. doi:10.1080/17483100802543114
- ^ a b c d e f g Wagner, K; Lechtzin, N; Judge, D (2007). "Current treatment of adult Duchenne muscular dystrophy". Biochimica et Biophysica Acta 1772 (2): 229–237. doi:10.1016/j.bbadis.2006.06.009. PMID 16887341
- ^ New England Journal of Medicine, 364;1513, Systemic administration of PRO051 in Duchenne's muscular dystrophy,
- ^ Study Shows Patients With Duchenne's Muscular Dystrophy Are Walking Better With PRO051 Treatment. By Daniel J. DeNoon WebMD Health News. March 23, 2011
- ^ a b England SB, Nicholson LV, Johnson MA, et al. (1990). "Very mild muscular dystrophy associated with the deletion of 46% of dystrophin". Nature 343 (6254): 180–2. doi:10.1038/343180a0. PMID 2404210.
- ^ Dominski Z, Kole R (1993). "Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides". Proc. Natl. Acad. Sci. U.S.A. 90 (18): 8673–7. doi:10.1073/pnas.90.18.8673. PMC 47420. PMID 8378346. http://www.pnas.org/cgi/pmidlookup?view=long&pmid=8378346.
- ^ Lacerra G, Sierakowska H, Carestia C, et al. (2000). "Restoration of hemoglobin A synthesis in erythroid cells from peripheral blood of thalassemic patients". Proc. Natl. Acad. Sci. U.S.A. 97 (17): 9591–6. doi:10.1073/pnas.97.17.9591. PMC 16909. PMID 10944225. http://www.pnas.org/cgi/pmidlookup?view=long&pmid=10944225.
- ^ Suwanmanee T, Sierakowska H, Lacerra G, et al. (2002). "Restoration of human beta-globin gene expression in murine and human IVS2-654 thalassemic erythroid cells by free uptake of antisense oligonucleotides". Mol. Pharmacol. 62 (3): 545–53. doi:10.1124/mol.62.3.545. PMID 12181431. http://molpharm.aspetjournals.org/cgi/pmidlookup?view=long&pmid=12181431.
- ^ Wilton SD, Lloyd F, Carville K, Fletcher S, Honeyman K, Agrawal S, Kole R (1999). "Specific removal of the nonsense mutation from the mdx dystrophin mRNA using antisense oligonucleotides". Neuromuscul Disord. 9 (5): 330–8. doi:10.1016/S0960-8966(99)00010-3. PMID 10407856.
- ^ Wilton SD, Fall AM, Harding PL, McClorey G, Coleman C, Fletcher S (2007). "Antisense oligonucleotide-induced exon skipping across the human dystrophin gene transcript". Mol. Ther. 15 (7): 1288–96. doi:10.1038/sj.mt.6300095. PMID 17285139.
- ^ Goyenvalle A, Vulin A, Fougerousse F, et al. (2004). "Rescue of dystrophic muscle through U7 snRNA-mediated exon skipping". Science 306 (5702): 1796–9. doi:10.1126/science.1104297. PMID 15528407.
- ^ Morgan JE, Pagel CN, Sherratt T, Partridge TA (1993). "Long-term persistence and migration of myogenic cells injected into pre-irradiated muscles of mdx mice". J. Neurol. Sci. 115 (2): 191–200. doi:10.1016/0022-510X(93)90224-M. PMID 7683332.
- ^ Dellavalle A, Sampaolesi M, Tonlorenzi R, et al. (2007). "Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells". Nat. Cell Biol. 9 (3): 255–67. doi:10.1038/ncb1542. PMID 17293855.
- ^ Common blood pressure drug treats muscular dystrophy in mice
- ^ Carre-Pierrat M, Mariol MC, Chambonnier L, et al. (2006). "Blocking of striated muscle degeneration by serotonin in C. elegans". J. Muscle Res. Cell. Motil. 27 (3–4): 253–8. doi:10.1007/s10974-006-9070-9. PMID 16791712.
- ^ Khairallah M, Khairallah RJ, Young ME, et al. (2008). "Sildenafil and cardiomyocyte-specific cGMP signaling prevent cardiomyopathic changes associated with dystrophin deficiency". Proc. Nat. Acad. Sci. U.S.A. 105 (19): 7028–33. doi:10.1073/pnas.0710595105. PMC 2383977. PMID 18474859. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2383977.
- ^ Louise R. Rodino-Klapac et al. (2007, Vol. 64, no. 9) "Gene Therapy for Duchenne Muscular Dystrophy". Arch. Neurol. Retrieved 1-10-2011
- ^ Researchers prove the gene responsible for Duchenne muscular dystrophy can be repaired, Published: Thursday, April 15, 2010 - 11:32 in Biology & Nature
- ^ Khurdayan VK, Bozzo J, Prous JR (Oct 2005). "Chronicles in drug discovery". Drug news & perspectives 18 (8): 517–522. doi:10.1358/dnp.2005.18.8.953409. ISSN 0214-0934. PMID 16391721.
- ^ MDA Research | Preliminary Results of DMD Clinical Trial Encouraging
- ^ http://www.parentprojectmd.org/site/DocServer/PTC124_PRESS_RELEASE.pdf?docID=1601
- ^ http://ptct.client.shareholder.com/releasedetail.cfm?ReleaseID=448803
- ^ ClinicalTrials.gov NCT00159250 Safety and Efficacy Study of Antisense Oligonucleotides in Duchenne Muscular Dystrophy
- ^ Clinical trial information for 2'-O-methyl phosphorothioate (PRO051) trial
- ^ http://www.genengnews.com/gen-news-highlights/shire-pays-acceleron-45m-up-front-for-neuromuscular-disease-candidates/81243899/
- ^ http://clinicaltrials.gov/ct2/show/NCT01099761
External links
- Dystrophy.com - General information about dystrophy treatments, organizations and causes
- Muscular Dystrophy - pediatric-orthopedics.com
- CDC’s National Center on Birth Defects and Developmental Disabilities (previously listed below as "Duchenne/Becker Muscular Dystrophy, NCBDDD") at CDC
- Online 'Mendelian Inheritance in Man' (OMIM) MUSCULAR DYSTROPHY -310200 on NCBI
- Genes and Disease Page at NCBI
- Muscular Dystrophies at the Open Directory Project
Muscular dystrophy The Nine Primary Muscular Dystrophies Congenital • dystrophin (Becker's, Duchenne) • Distal • Emery-Dreifuss • Facioscapulohumeral • Limb-girdle muscular dystrophy • Myotonic • OculopharyngealRelated topicsNational/International Organizations US government Institutes and Legislation National/International Events MDA Labor Day Telethon (USA) • Décrypthon (France)Recent or Ongoing Clinical Trials Diseases of myoneural junction and muscle / neuromuscular disease (G70–G73, 358–359) Neuromuscular-
junction diseaseautoimmune (Myasthenia gravis, Lambert–Eaton myasthenic syndrome)Myopathy/
congenital myopathyMuscular dystrophy
(DAPC)ADAROther structuralOtherOtherCategories:- Muscular dystrophy
- X-linked recessive disorders
Wikimedia Foundation. 2010.