- Chronic granulomatous disease
-
Chronic granulomatous disease Classification and external resources 
SuperoxideICD-10 D71 ICD-9 288.1 OMIM 306400 233690 233700 DiseasesDB 2633 MedlinePlus 001239 eMedicine ped/1590 derm/719 MeSH D006105 Chronic granulomatous disease (CGD) (also known as Bridges–Good syndrome, Chronic granulomatous disorder, and Quie syndrome[1]) is a diverse group of hereditary diseases in which certain cells of the immune system have difficulty forming the reactive oxygen compounds (most importantly, the superoxide radical) used to kill certain ingested pathogens.[2] This leads to the formation of granulomata in many organs.[3] CGD affects about 1 in 200,000 people in the United States, with about 20 new cases diagnosed each year.[4][5]
This condition was first discovered in 1950 in a series of 4 boys from Minnesota, and in 1957 was named "a fatal granulomatosus of childhood" in a publication describing their disease.[6][7] The underlying cellular mechanism that causes chronic granulomatous disease was discovered in 1967, and research since that time has further elucidated the molecular mechanisms underlying the disease.[8] In 1986, the x-linked form of CGD was the first disease for which positional cloning was used to identify the underlying genetic mutation.
Contents
Classification
Chronic granulomatous disease is the name for a genetically heterogeneous group of immunodeficiencies. The core defect is a failure of phagocytic cells to kill organisms that they have engulfed because of defects in a system of enzymes that produce free radicals and other toxic small molecules. There are several types, including chronic X-linked disease, chronic b-negative disease, X-linked cytochrome b-positive disease, x-linked variant disease, and atypical granulomatous disease.[9]
Symptoms
Classically, patients with chronic granulomatous disease will suffer from recurrent bouts of infection due to the decreased capacity of their immune system to fight off disease-causing organisms. The recurrent infections they acquire are specific and are, in decreasing order of frequency:
- pneumonia
- abscesses of the skin, tissues, and organs
- suppurative arthritis
- osteomyelitis
- bacteremia/fungemia
- superficial skin infections such as cellulitis or impetigo
Most people with CGD are diagnosed in childhood, usually before age 5.[10] Early diagnosis is important since these people can be placed on antibiotics to ward off infections before they occur.
Atypical infections
 Microscopic image of the fungus, Aspergillus fumigatus, an organism that commonly causes disease in people with chronic granulomatous disease.
Microscopic image of the fungus, Aspergillus fumigatus, an organism that commonly causes disease in people with chronic granulomatous disease.
People with CGD are sometimes infected with organisms that usually do not cause disease in people with normal immune systems. Among the most common organisms that cause disease in CGD patients are:
- bacteria (particularly those that are catalase-positive)[11]
- Staphylococcus aureus.
- Serratia marcescens.
- Salmonella species.
- Klebsiella species.
- Pseudomonas cepacia, a.k.a. Burkholderia cepacia.[12]
- Nocardia.[13]
- fungi
- Aspergillus species. Aspergillus has a propensity to cause infection in people with CGD and of the Aspergillus species, Aspergillus fumigatus seems to be most common in CGD.
- Candida species.
Patients with CGD can usually resist infections of catalase-negative bacteria. Catalase is an enzyme that catalyzes the breakdown of hydrogen peroxide in many organisms. In organisms that lack catalase (catalase-negative), normal metabolic functions will cause an accumulation of hydrogen peroxide which the host's immune system can use to fight off the infection. In organisms that have catalase (catalase-positive), the enzyme breaks down any hydrogen peroxide that was produced through normal metabolism. Therefore hydrogen peroxide will not accumulate, leaving the patient vulnerable to catalase-positive bacteria.
Genetics
Most cases of chronic granulomatous disease are transmitted as a mutation on the X chromosome and are thus called an "X-linked trait".[10] The affected gene on the X chromosome codes for the gp91 protein p91-PHOX (p is the weight of the protein in kDa; the g means glycoprotein). CGD can also be transmitted in an autosomal recessive fashion (via CYBA and NCF1) and affects other PHOX proteins. The type of mutation that causes both types of CGD are varied and may be deletions, frame-shift, nonsense, and missense.[14][15]
A low level of NADPH, the cofactor required for superoxide synthesis, can lead to CGD. This has been reported in women who are homozygous for the genetic defect causing glucose-6-phosphate dehydrogenase deficiency (G6PD), which is characterised by reduced NADPH levels.[citation needed]
Pathophysiology
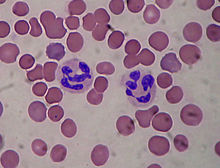 Two neutrophils among many red blood cells. Neutrophils are one type of cell affected by chronic granulomatous disease.
Two neutrophils among many red blood cells. Neutrophils are one type of cell affected by chronic granulomatous disease.Phagocytes (i.e., neutrophils, monocytes, and macrophages) require an enzyme to produce reactive oxygen species to destroy bacteria after they ingest the bacteria in a process called phagocytosis, a process known as the respiratory burst. This enzyme is termed "phagocyte NADPH oxidase" (PHOX). The initial step in this process involves the one-electron reduction of molecular oxygen to produce superoxide anion, a free radical. Superoxide then undergoes a further series of reactions to produce products such as hydrogen peroxide (through the action of superoxide dismutase), hydroxyl radical and hypochlorite (bleach - through the action of myeloperoxidase on hydrogen peroxide). The reactive oxygen species this enzyme produces are toxic to bacteria and help the phagocyte kill them once they are ingested. Defects in one of the four essential subunits of this enzyme can all cause CGD of varying severity, dependent on the defect. There are over 410 known possible defects in the PHOX enzyme complex that can lead to chronic granulomatous disease.[3]
Diagnosis
The nitroblue-tetrazolium (NBT) test is the original and most widely-known test for chronic granulomatous disease.[16] It is positive in CGD, meaning that it does not turn blue (a "positive" test result indicates that the disease in question is present). The higher the blue score, the better the cell is at producing reactive oxygen species. This test depends upon the direct reduction of NBT by superoxide free radical to form an insoluble formazan. This test is simple to perform and gives rapid results, but only tells whether or not there is a problem with the PHOX enzymes, not how much they are affected. A similar test uses dihydrorhodamine (DHR) where whole blood is stained with DHR, incubated, and stimulated to produce superoxide radicals which reduce DHR to rhodamin in cells with normal function. An advanced test called the cytochrome C reduction assay tells physicians how much superoxide a patient's phagocytes can produce. Once the diagnosis of CGD is established, a genetic analysis may be used to determine exactly which mutation is the underlying cause.[citation needed]
Treatment
Management of chronic granulomatous disease revolves around two goals: 1) diagnose the disease early so that antibiotic prophylaxis can be given to keep an infection from occurring, and 2) educate the patient about his or her condition so that prompt treatment can be given if an infection occurs.[citation needed]
Antibiotics
Physicians often prescribe the antibiotic trimethoprim-sulfamethoxazole to prevent bacterial infections.[17] This drug also has the benefit of sparing the normal bacteria of the digestive tract. Fungal infection is commonly prevented with itraconazole,[18] although a newer drug of the same type called voriconazole may be more effective.[19] The use of this drug for this purpose is still under scientific investigation.
Immunomodulation
Interferon, in the form of interferon gamma-1b (Actimmune) is approved by the Food and Drug Administration for the prevention of infection in CGD. It has been shown to prevent infections in CGD patients by 70% and to reduce their severity. Although its exact mechanism is still not entirely understood, it has the ability to give CGD patients more immune function and therefore, greater ability to fight off infections. This therapy has been standard treatment for CGD for several years.[20]
Hematopoietic stem cell transplantation (HSCT)
Hematopoietic stem cell transplantation from a matched donor is curative although not without significant risk.[21][22]
Prognosis
There are currently no studies detailing the long term outcome of chronic granulomatous disease with modern treatment. Without treatment, children often die in the first decade of life. The increased severity of X-linked CGD results in a decreased survival rate of patients, as 20% of X-link patients die of CGD-related causes by the age of 10, in contrast to an approximate age of 35 in autosomal recessive patients.[23]
Epidemiology
CGD affects about 1 in 200,000 people in the United States, with about 20 new cases diagnosed each year.[4][5]
Chronic granulomatous disease affects all people of all races, however, there is limited information on prevalence outside of the United States. One survey in Sweden reported an incidence of 1 in 220,000 people,[24], while a larger review of studies in Europe suggested a larger 1 in 250,000 people.[23]
History
This condition was first described in 1954 by Janeway, who reported five cases of the disease in children.[25] In 1957 it was further characterized as "a fatal granulomatosus of childhood".[6][7] The underlying cellular mechanism that causes chronic granulomatous disease was discovered in 1967, and research since that time has further elucidated the molecular mechanisms underlying the disease.[8] Use of antibiotic prophylaxis, surgical abscess drainage, and vaccination lead to the term "fatal" being dropped from the name of the disease as children survived into adulthood. The oldest person to suffer from Chronic Granulomatos Disease was Mr. Jackie Ray Johnson of Fredericksburg, Virginia who died in 2002 at the age of 63.
Research
Gene therapy is currently being studied as a possible treatment for chronic granulomatous disease. CGD is well-suited for gene therapy since it is caused by a mutation in single gene which only affects one body system (the hematopoietic system). Viruses have been used to deliver a normal gp91 gene to rats with a mutation in this gene, and subsequently the phagocytes in these rats were able to produce oxygen radicals.[26]
In 2006, two human patients with X-linked chronic granulomatous disease underwent gene therapy and blood cell precursor stem cell transplantation to their bone marrow. Both patients recovered from their CGD, clearing pre-existing infections and demonstrating increased oxidase activity in their neutrophils. However, long-term complications and efficacy of this therapy are unknown.[27]
References
- ^ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. ISBN 1-4160-2999-0.
- ^ "Chronic Granulomatous Disease: Immunodeficiency Disorders: Merck Manual Professional". http://www.merck.com/mmpe/sec13/ch164/ch164e.html. Retrieved 2008-03-01.
- ^ a b Heyworth P, Cross A, Curnutte J (2003). "Chronic granulomatous disease". Curr Opin Immunol 15 (5): 578–84. doi:10.1016/S0952-7915(03)00109-2. PMID 14499268.
- ^ a b Maryland Pao, M.D. et al. (2004). "Cognitive Function in Patients With Chronic Granulomatous Disease: A Preliminary Report". Psychosomatics 45 (June 2004): 230–4. doi:10.1176/appi.psy.45.3.230. PMID 15123849.
- ^ a b James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. ISBN 0-7216-2921-0.
- ^ a b Berendes H, Bridges RA, Good RA (1957). "A fatal granulomatosus of childhood: the clinical study of a new syndrome". Minn Med 40 (5): 309–12. PMID 13430573.
- ^ a b Bridges RA, Berendes H, Good RA (1959). "A fatal granulomatous disease of childhood; the clinical, pathological, and laboratory features of a new syndrome". AMA J Dis Child 97 (4): 387–408. PMID 13636694.
- ^ a b Baehner RL, Nathan DG (1967). "Leukocyte oxidase: defective activity in chronic granulomatous disease". Science 155 (3764): 835–6. doi:10.1126/science.155.3764.835. PMID 6018195.
- ^ http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=306400
- ^ a b Winkelstein J, Marino M, Johnston R, Boyle J, Curnutte J, Gallin J, Malech H, Holland S, Ochs H, Quie P, Buckley R, Foster C, Chanock S, Dickler H (2000). "Chronic granulomatous disease. Report on a national registry of 368 patients". Medicine (Baltimore) 79 (3): 155–69. doi:10.1097/00005792-200005000-00003. PMID 10844935.
- ^ Soler-Palacín P, Margareto C, Llobet P, et al. (2007). "Chronic granulomatous disease in pediatric patients: 25 years of experience". Allergol Immunopathol (Madr) 35 (3): 83–9. doi:10.1157/13106774. PMID 17594870. http://www.elsevier.es/revistas/0301-0546/35/83. Retrieved 2009-04-08.
- ^ Lacy DE, Spencer DA, Goldstein A, Weller PH, Darbyshire P (November 1993). "Chronic granulomatous disease presenting in childhood with Pseudomonas cepacia septicaemia". J. Infect. 27 (3): 301–4. doi:10.1016/0163-4453(93)92271-W. PMID 7508484. http://linkinghub.elsevier.com/retrieve/pii/0163-4453(93)92271-W. Retrieved 2009-04-08.
- ^ Dorman SE, Guide SV, Conville PS, et al. (August 2002). "Nocardia infection in chronic granulomatous disease". Clin. Infect. Dis. 35 (4): 390–4. doi:10.1086/341416. PMID 12145721.
- ^ Heyworth P, Curnutte J, Rae J, Noack D, Roos D, van Koppen E, Cross A (2001). "Hematologically important mutations: X-linked chronic granulomatous disease (second update)". Blood Cells Mol Dis 27 (1): 16–26. doi:10.1006/bcmd.2000.0347. PMID 11162142.
- ^ Cross A, Noack D, Rae J, Curnutte J, Heyworth P (2000). "Hematologically important mutations: the autosomal recessive forms of chronic granulomatous disease (first update)". Blood Cells Mol Dis 26 (5): 561–5. doi:10.1006/bcmd.2000.0333. PMID 11112388.
- ^ Kasper, DL, Fauci, AS, Longo, DL, Braunwald, E, Hauser, SL, and Jameson, JL. Harrison's Principles of Internal Medicine, 16th edition 2005;357. ISBN 0-07-139140-1.
- ^ Weening R, Kabel P, Pijman P, Roos D (1983). "Continuous therapy with sulfamethoxazole-trimethoprim in patients with chronic granulomatous disease". J Pediatr 103 (1): 127–30. doi:10.1016/S0022-3476(83)80798-7. PMID 6408232.
- ^ Cale C, Jones A, Goldblatt D (2000). "Follow up of patients with chronic granulomatous disease diagnosed since 1990". Clin Exp Immunol 120 (2): 351–5. doi:10.1046/j.1365-2249.2000.01234.x. PMC 1905649. PMID 10792387. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1905649.
- ^ Sabo J, Abdel-Rahman S (2000). "Voriconazole: a new triazole antifungal". Ann Pharmacother 34 (9): 1032–43. doi:10.1345/aph.19237. PMID 10981251.
- ^ "A controlled trial of interferon gamma to prevent infection in chronic granulomatous disease. The International Chronic Granulomatous Disease Cooperative Study Group". N Engl J Med 324 (8): 509–16. 1991. doi:10.1056/NEJM199102213240801. PMID 1846940.
- ^ Jones LB, McGrogan P, Flood TJ, et al. (May 2008). "Special article: chronic granulomatous disease in the United Kingdom and Ireland: a comprehensive national patient-based registry". Clin. Exp. Immunol. 152 (2): 211–8. doi:10.1111/j.1365-2249.2008.03644.x. PMC 2384093. PMID 18410635. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2384093.
- ^ Soncini E, Slatter MA, Jones LB, et al. (April 2009). "Unrelated donor and HLA-identical sibling haematopoietic stem cell transplantation cure chronic granulomatous disease with good long-term outcome and growth". Br. J. Haematol. 145 (1): 73–83. doi:10.1111/j.1365-2141.2009.07614.x. PMID 19222467.
- ^ a b van den Berg MJ, Van Koppen E, Ahlin A, et al. (2009). Alspaugh, Andy. ed. "Chronic Granulomatous Disease: The European Experience". PLoS One 4 (4): e5234. doi:10.1371/journal.pone.0005234. PMC 2668749. PMID 19381301. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2668749.
- ^ http://emedicine.medscape.com/article/956936-overview
- ^ http://www.medimmunol.com/content/5/1/4#B1
- ^ Dinauer M, Gifford M, Pech N, Li L, Emshwiller P (2001). "Variable correction of host defense following gene transfer and bone marrow transplantation in murine X-linked chronic granulomatous disease". Blood 97 (12): 3738–45. doi:10.1182/blood.V97.12.3738. PMID 11389011.
- ^ Ott M, Schmidt M, Schwarzwaelder K, et al. (2006). "Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1". Nat Med 12 (4): 401–9. doi:10.1038/nm1393. PMID 16582916.
External links
- The Chronic Granulomatous Disease Association
- Chronic Granulomatous Disorder Research Trust
- WASOG - World Association of Sarcoidosis and Other Granulomatous Disorders
Hematologic disease: Monocyte and granulocyte disease (CFU-GM/CFU-Baso/CFU-Eos), including immunodeficiency (D70-D71, 288) Monocytes/
macrophages↑↓Granulocytes ↑↓PBD chemotaxis/degranulation: Leukocyte adhesion deficiency (LAD1, LAD2) · Chédiak–Higashi syndrome
respiratory burst: (Chronic granulomatous disease, Neutrophil immunodeficiency syndrome, Myeloperoxidase deficiency)Categories:- Congenital defects of phagocyte number, function, or both
- Enzyme defects
- Noninfectious immunodeficiency-related cutaneous conditions


Wikimedia Foundation. 2010.