- Dent's disease
-
Dent's disease Classification and external resources 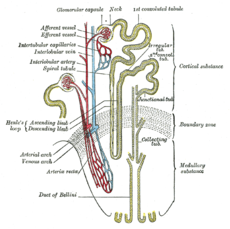
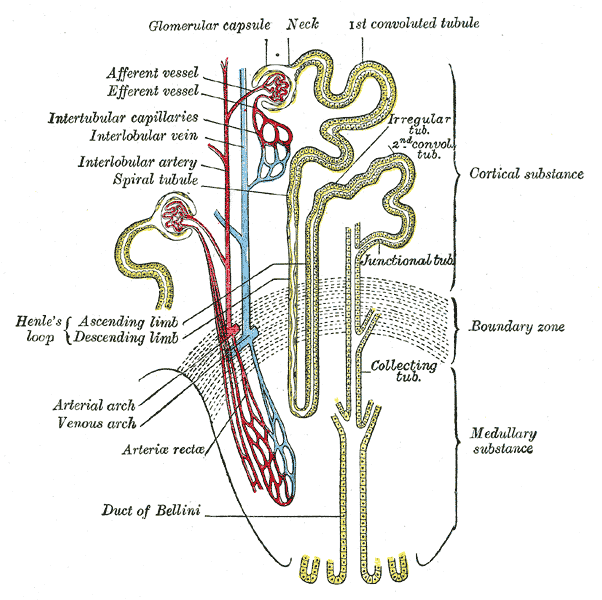
Nephron of the kidney without juxtaglomerular apparatusOMIM 300009 DiseasesDB 29911 Dent's disease (or Dent disease) is a rare X-linked recessive inherited condition that affects the proximal renal tubules[1] of the kidney. It is one cause of Fanconi syndrome, and is characterized by tubular proteinuria, hypercalciuria, calcium nephrolithiasis, nephrocalcinosis and chronic renal failure.
"Dent's disease" is often used to describe an entire group of familial disorders, including X-linked recessive nephrolithiasis with renal failure, X-linked recessive hypophosphataemic rickets, and both Japanese and idiopathic low molecular weight proteinuria.[2]
Contents
History
Dent's disease was first described by Dent, C. E. and Friedman, M in 1964 when they reported 2 unrelated British boys with rickets associated with renal tubular damage characterized by hypercalciuria, hyperphosphaturia, proteinuria, and aminoaciduria.[3] This is a genetic disorder caused by the genetic mutations in the renal chloride channel CLCN5 which encodes a kidney-specific voltage gated chloride channel and a 746 amino acid protein (CLC-5), with 12 to 13 transmembrane domains; it manifests itself through low molecular weight proteinuria, hypercalciuria, aminoaciduria and hypophosphataemia. Because of its rather rare occurrence, Dent's disease is often diagnosed as idiopathic hypercalciuria (IH), i.e. excess calcium in urine with undetermined causes.
Genetics
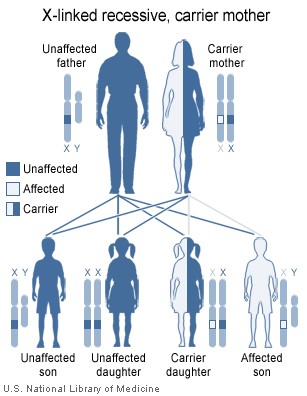
 X-linked recessive inheritance.
X-linked recessive inheritance.
Dent disease 1
Dent's disease is a X-linked recessive disorder. The males are prone to manifesting symptoms in early adulthood with symptoms of calculi, rickets or even with renal failure in more severe cases.
In humans, gene CLCN5 is located on chromosome Xp11.22 and has a 2238-bp coding sequence that consists of 11 exons that span 25 to 30 kb of genomic DNA and encode a 746 amino acid protein.[4] CLCN5 belongs to the family of voltage-gated chloride channel genes (CLCN1-CLCN7, and CLCKa and CLCKb) that have approximately 12 transmembrane domains. These chloride channels have an important role in the control of membrane excitability, transepithelial transport, and possibly cell volume.[5]
The mechanisms by which CLC-5 dysfunction results in hypercalciuria and the other features of Dent's disease remain to be elucidated. The identification of additional CLCN5 mutations may help in these studies, and we have pursued such studies in patients with Dent's disease.[6]
Dent disease 2
Dent disease 2 is associated with OCRL.[7][8]
Symptoms
Dent's disease often produces symptoms of:
- Extreme thirst combined with dehydration which leads to frequent urination
- Nephrolithiasis (kidney stones)
- Hypercalciuria (high urine calcium - >300 mg/d or >4 mg/kg per d) with normal levels blood/serum calcium)
Dent's disease may also be associated with:
- Aminoaciduria (amino acids in urine)
- Phosphaturia (phosphate in urine)
- Glycosuria (glucose in urine)
- Kaliuresis (potassium in urine)
- Hyperuricosuria (excessive amounts of uric acid in the urine.)
- Impaired urinary acidification
- Rickets
In a very large study of patients with Dent's disease, 9 out of 15 men, and 1 out of 10 women suffered end-stage renal failure by the age of 47.[9]
Treatment
As of today, there is no agreed-upon treatment of Dent's disease and no therapy has been formally accepted. Most treatment measures are mostly supportive in nature and they include:
- Thiazide diuretics (i.e. Hydrochlorothiazide) which have been used with success in reducing the calcium output in urine, but they are also known to cause hypokalemia.
- In rats with diabetes insipidus thiazide diuretics inhibit the NaCl co-transporter in the renal distal convoluted tubule leading indirectly to less water and solutes being delivered to the distal tubule.[10] The impairment of Na transport in the distal convoluted tubule, induces natriuresis and water loss while increasing the reabsorption of calcium in this segment in a manner unrelated to sodium transport.
- Amiloride which also increases distal tubular calcium reabsorption and has been used as a therapy for idiopathic hypercalciuria.
- A combination of 25 mg of chlorthalidone plus 5 mg of amiloride daily led to a substantial reduction in urine calcium in Dent's patients, however urine pH was "significantly higher in patients with Dent’s disease than in those with idiopathic hypercalciuria (P < 0.03), and supersaturation for uric acid was consequently lower (P < 0.03)."[11]
- For patients with osteomalacia, Vitamin D or derivatives have been employed, apparently with success.
- Some lab tests on mice with CLC-5 related tubular damage showed that a high citrate diet preserved renal function and delayed progress of renal disease.[12]
External links
References
- ^ "Dent disease" at Dorland's Medical Dictionary
- ^ Mayo Clinic, Division of Nephrology and Hypertension, Mineral Metabolism and Stone Disease
- ^ Dent CE, Friedman M. Hypercalciuric rickets associated with renal tubular damage
- ^ Fisher SE, van Bakel I, Lloyd SE, Pearce SH, Thakker RV, Craig IW (October 1995). "Cloning and characterization of CLCN5, the human kidney chloride channel gene implicated in Dent disease (an X-linked hereditary nephrolithiasis)". Genomics 29 (3): 598–606. doi:10.1006/geno.1995.9960. PMID 8575751. http://linkinghub.elsevier.com/retrieve/pii/S0888754385799600.
- ^ Jentsch TJ, Friedrich T, Schriever A, Yamada H (May 1999). "The CLC chloride channel family". Pflugers Arch. 437 (6): 783–95. doi:10.1007/s004240050847. PMID 10370055. http://link.springer.de/link/service/journals/00424/bibs/9437006/94370783.htm.
- ^ Yamamoto K, Cox JP, Friedrich T, et al. (August 2000). "Characterization of renal chloride channel (CLCN5) mutations in Dent's disease". J. Am. Soc. Nephrol. 11 (8): 1460–8. PMID 10906159. http://jasn.asnjournals.org/cgi/content/full/11/8/1460.
- ^ Online 'Mendelian Inheritance in Man' (OMIM) 300555
- ^ Hoopes RR, Shrimpton AE, Knohl SJ, et al. (February 2005). "Dent Disease with mutations in OCRL1". Am. J. Hum. Genet. 76 (2): 260–7. doi:10.1086/427887. PMC 1196371. PMID 15627218. http://linkinghub.elsevier.com/retrieve/pii/S0002-9297(07)62577-4.
- ^ Burgess HK, Jayawardene SA, Velasco N (July 2001). "Dent's disease: can we slow its progression?". Nephrol. Dial. Transplant. 16 (7): 1512–3. doi:10.1093/ndt/16.7.1512. PMID 11427657. http://ndt.oxfordjournals.org/cgi/content/full/16/7/1512.
- ^ Loffing J (November 2004). "Paradoxical antidiuretic effect of thiazides in diabetes insipidus: another piece in the puzzle". J. Am. Soc. Nephrol. 15 (11): 2948–50. doi:10.1097/01.ASN.0000146568.82353.04. PMID 15504949. http://jasn.asnjournals.org/cgi/content/full/15/11/2948.
- ^ Raja KA, Schurman S, D'mello RG, et al. (December 2002). "Responsiveness of hypercalciuria to thiazide in Dent's disease". J. Am. Soc. Nephrol. 13 (12): 2938–44. doi:10.1097/01.ASN.0000036869.82685.F6. PMID 12444212. http://jasn.asnjournals.org/cgi/content/full/13/12/2938.
- ^ Cebotaru V, Kaul S, Devuyst O, et al. (August 2005). "High citrate diet delays progression of renal insufficiency in the ClC-5 knockout mouse model of Dent's disease". Kidney Int. 68 (2): 642–52. doi:10.1111/j.1523-1755.2005.00442.x. PMID 16014041.
Congenital malformations and deformations of urinary system (Q60–Q64, 753) Abdominal KidneyPelvic Vestigial Genetic disorder, membrane: Channelopathy Calcium channel CACNA1A (Familial hemiplegic migraine 1, Episodic ataxia 2, Spinocerebellar ataxia type-6) · CACNA1C (Timothy syndrome, Brugada syndrome 3, Long QT syndrome 8) · CACNA1F (Ocular albinism 2, CSNB2A) · CACNA1S (Hypokalemic periodic paralysis 1, Thyrotoxic periodic paralysis 1) · CACNB2 (Brugada syndrome 4)Ligand gatedSodium channel Voltage-gatedSCN1A (Familial hemiplegic migraine 3, GEFS+ 2, Febrile seizure 3A) · SCN1B (Brugada syndrome 6, GEFS+ 1) · SCN4A (Hypokalemic periodic paralysis 2, Hyperkalemic periodic paralysis, Paramyotonia congenita, Potassium-aggravated myotonia) · SCN4B (Long QT syndrome 10) · SCN5A (Brugada syndrome 1, Long QT syndrome 3) · SCN9A (Erythromelalgia, Febrile seizure 3B, Paroxysmal extreme pain disorder, Congenital insensitivity to pain)Potassium channel KCNA1 (Episodic ataxia 1) · KCNA5 (Familial atrial fibrillation 7) · KCNC3 (Spinocerebellar ataxia type-13) · KCNE1 (Jervell and Lange-Nielsen syndrome, Long QT syndrome 5) · KCNE2 (Long QT syndrome 6) · KCNE3 (Brugada syndrome 5) · KCNH2 (Short QT syndrome) · KCNQ1 (Jervell and Lange-Nielsen syndrome, Romano-Ward syndrome, Short QT syndrome, Long QT syndrome 1, Familial atrial fibrillation 3) · KCNQ2 (BFNS1}KCNJ1 (Bartter syndrome 2) · KCNJ2 (Andersen-Tawil syndrome, Long QT syndrome 7, Short QT syndrome) · KCNJ11 (TNDM3) · KCNJ18 (Thyrotoxic periodic paralysis 2)Chloride channel CFTR (Cystic fibrosis, Congenital absence of the vas deferens) · CLCN1 (Thomsen disease, Myotonia congenita) · CLCN5 (Dent's disease) · CLCN7 (Osteopetrosis A2, B4 · BEST1 (Vitelliform macular dystrophy) · CLCNKB (Bartter syndrome 3)TRP channel Connexin GJA1 (Oculodentodigital dysplasia, Hallermann–Streiff syndrome, Hypoplastic left heart syndrome) · GJB1 (Charcot–Marie–Tooth disease X1) · GJB2 (Keratitis–ichthyosis–deafness syndrome, Ichthyosis hystrix, Bart–Pumphrey syndrome, Vohwinkel syndrome) · GJB3/GJB4 (Erythrokeratodermia variabilis, Progressive symmetric erythrokeratodermia) · GJB6 (Clouston's hidrotic ectodermal dysplasia)Porin see also ion channels
B structural (perx, skel, cili, mito, nucl, sclr) · DNA/RNA/protein synthesis (drep, trfc, tscr, tltn) · membrane (icha, slcr, atpa, abct, othr) · transduction (iter, csrc, itra), trfkCategories:- Channelopathy
- Kidney diseases
- Congenital disorders of urinary system
- Rare diseases
Wikimedia Foundation. 2010.