- Arrhythmogenic right ventricular dysplasia
-
Arrhythmogenic right ventricular dysplasia Classification and external resources 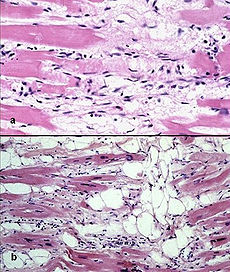
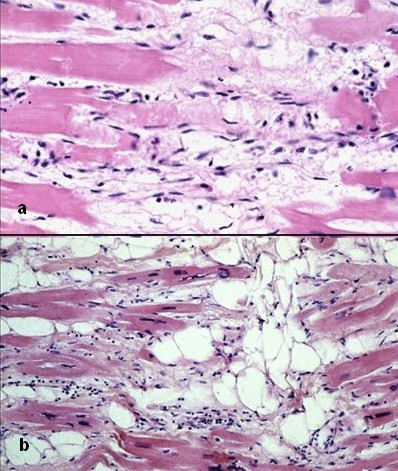
Photomicrograph of an ARVC heart.ICD-10 I42.8 OMIM 107970 DiseasesDB 29750 MeSH D019571 Arrhythmogenic right ventricular dysplasia (ARVD), also called arrhythmogenic right ventricular cardiomyopathy (ARVC) or arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C), is an inherited heart disease.
ARVD is caused by genetic defects of the parts of heart muscle (also called myocardium or cardiac muscle) known as desmosomes, areas on the surface of heart muscle cells which link the cells together. The desmosomes are composed of several proteins, and many of those proteins can have harmful mutations.
The disease is a type of nonischemic cardiomyopathy that involves primarily the right ventricle. It is characterized by hypokinetic areas involving the free wall of the right ventricle, with fibrofatty replacement of the right ventricular myocardium, with associated arrhythmias originating in the right ventricle.
ARVD is often found in association with diffuse palmoplantar keratoderma, and woolly hair, because their genes are nearby and often inherited together [1]:513[2]
ARVC/D is an important cause of ventricular arrhythmias in children and young adults. It is seen predominantly in males, and 30-50% of cases have a familial distribution.
Contents
Genetics
It is usually inherited in an autosomal dominant pattern, with variable expression. Novel studies showed that mutations (point mutations) in genes encoding for desmosomal proteins (see Intercalated Disc) are the main causatives for the development of this disease. Recently it could be shown, that mutations in the desmin-gene could cause ARVC. Desmin is a intermediate filament protein, which is linked to the desmosomes. The penetrance is 20-35% in general, but significantly higher in Italy. Seven gene loci have been implicated in ARVD. However, about 50% of families that express ARVD that undergo genetic screening do not show linkage with any of the known chromosomal loci. It is unclear whether the pathogenesis varies with the different loci involved. Standard genetic screening test are currently tested and evaluated in different state of the art cardiovascular research centres and hospitals. Types include:
Type OMIM Gene Locus ARVD1 107970 TGFB3 14q23-q24 ARVD2 600996 RYR2 1q42-q43 ARVD3 602086 ? 14q12-q22 ARVD4 602087 ? 2q32.1-q32.3 ARVD5 604400 TMEM43 3p23 ARVD6 604401 ? 10p14-p12 ARVD7 609160 ? 10q22.3 ARVD8 607450 DSP 6p24 ARVD9 609040 PKP2 12p11 ARVD10 610193 DSG2 18q12.1-q12 ARVD11 610476 DSC2 18q12.1 ARVD12 611528 JUP 17q21 Incidence
The incidence of ARVD is about 1/10,000 in the general population in the United States, although some studies have suggested that it may be as common as 1/1,000. Recently, 1/200 were found to be carriers of mutations that predispose to ARVC[3] It accounts for up to 17% of all sudden cardiac deaths in the young. In Italy, the incidence is 40/10,000, making it the most common cause of sudden cardiac death in the young population.
Presentation
Up to 80% of individuals with ARVD present with syncope or sudden cardiac death. The remainder frequently present with palpitations or other symptoms due to right ventricular outflow tract (RVOT) tachycardia (a type of monomorphic ventricular tachycardia).
Symptoms are usually exercise-related. In populations where hypertrophic cardiomyopathy is screened out prior to involvement in competitive athletics, it is a common cause of sudden cardiac death.
The first clinical signs of ARVD are usually during adolescence. However, signs of ARVD have been demonstrated in infants.
Pathogenesis
The pathogenesis of ARVD is largely unknown. Apoptosis (programmed cell death) appears to play a large role. It is unclear why only the right ventricle is involved. The disease process starts in the subepicardial region and works its way towards the endocardial surface, leading to transmural involvement (possibly accounting for the aneurysmal dilatation of the RV). Residual myocardium is confined to the subendocardial region and the trabeculae of the RV. These trabeculae may become hypertrophied.
Aneurysmal dilatation is seen in 50% of cases at autopsy. It usually occurs in the diaphragmatic, apical, and infundibular regions (known as the triangle of dysplasia). The left ventricle is involved in 50-67% of individuals. If the left ventricle is involved, it is usually late in the course of disease, and confers a poor prognosis.
There are two pathological patterns seen in ARVD, Fatty infiltration and fibro-fatty infiltration.
Fatty infiltration
The first, fatty infiltration, is confined to the right ventricle. This involves a partial or near-complete substitution of myocardium with fatty tissue without wall thinning. It involves predominantly the apical and infundibular regions of the RV. The left ventricle and ventricular septum are usually spared. No inflammatory infiltrates are seen in fatty infiltration. There is evidence of myocyte (myocardial cell) degeneration and death seen in 50% of cases of fatty infiltration.
Fibro-fatty infiltration
The second, fibro-fatty infiltration, involves replacement of myocytes with fibrofatty tissue. A patchy myocarditis is involved in up to 2/3 of cases, with inflammatory infiltrates (mostly T cells) seen on microscopy. Myocardial atrophy is due to injury and apoptosis. This leads to thinning of the RV free wall (to < 3 mm thickness) Myocytes are replaced with fibrofatty tissue. The regions preferentially involved include the RV inflow tract, the RV outflow tract, and the RV apex. However, the LV free wall may be involved in some cases. Involvement of the ventricular septum is rare. The areas involved are prone to aneurysm formation.
Ventricular arrhythmias
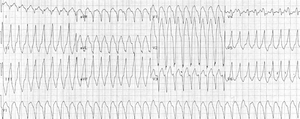
Right ventricular outflow tract tachycardia
Monomorphic ventricular tachycardia originating from the right ventricular outflow tract. Ventricular arrhythmias due to ARVD typically arise from the diseased right ventricle. The type of arrhythmia ranges from frequent premature ventricular complexes (PVCs) to ventricular tachycardia (VT) to ventricular fibrillation (VF).
While the initiating factor of the ventricular arrhythmias is unclear, it may be due to triggered activity or reentry.
Ventricular arrhythmias are usually exercise-related, suggesting that they are sensitive to catecholamines. The ventricular beats typically have a right axis deviation. Multiple morphologies of ventricular tachycardia may be present in the same individual, suggesting multiple arrhythmogenic foci or pathways.
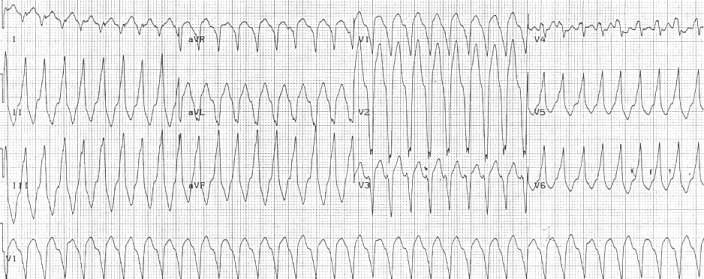
Right ventricular outflow tract (RVOT) tachycardia is the most common VT seen in individuals with ARVD. In this case, the EKG shows a left bundle branch block (LBBB) morphology with an inferior axis.
Diagnosis
The differential diagnosis for the ventricular tachycardia due to ARVD include:
- Congenital heart disease
- Repaired tetralogy of Fallot
- Ebstein's anomaly
- Uhl's anomaly
- Atrial septal defect
- Partial anomalous venous return
- Acquired heart disease
- Tricuspid valve disease
- Pulmonary hypertension
- Right ventricular infarction
- Bundle-branch re-entrant tachycardia
- Miscellaneous
- Pre-excited AV re-entry tachycardia
- Idiopathic RVOT tachycardia
- Sarcoidosis
Clinical testing
In order to make the diagnosis of ARVD, a number of clinical tests are employed, including the electrocardiogram (EKG), echocardiography, right ventricular angiography, cardiac MRI, and genetic testing.
Electrocardiogram
90% of individuals with ARVD have some EKG abnormality. The most common EKG abnormality seen in ARVD is T wave inversion in leads V1 to V3. However, this is a non-specific finding, and may be considered a normal variant in right bundle branch block (RBBB), women, and children under 12 years old.
RBBB itself is seen frequently in individuals with ARVD. This may be due to delayed activation of the right ventricle, rather than any intrinsic abnormality in the right bundle branch.
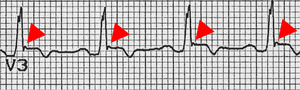
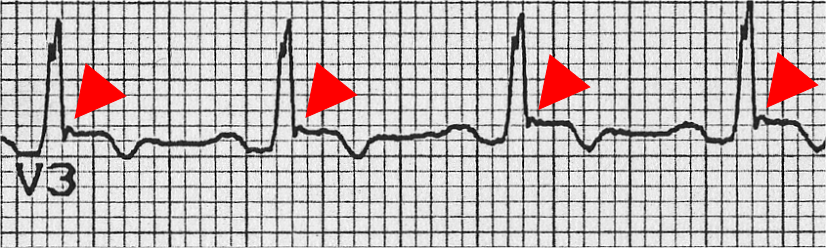
The epsilon wave
The epsilon wave (marked by red triangle), seen in ARVD. The epsilon wave is found in about 50% of those with ARVD. This is described as a terminal notch in the QRS complex. It is due to slowed intraventricular conduction. The epsilon wave may be seen on a surface EKG; however, it is more commonly seen on signal averaged EKGs.
Ventricular ectopy seen on a surface EKG in the setting of ARVD is typically of left bundle branch block (LBBB) morphology, with a QRS axis of -90 to +110 degrees. The origin of the ectopic beats is usually from one of the three regions of fatty degeneration (the "triangle of dysplasia"): the RV outflow tract, the RV inflow tract, and the RV apex.
Signal averaged ECG
Signal averaged ECG (SAECG) is used to detect late potentials and epsilon waves in individuals with ARVD.
Echocardiography
Echocardiography may reveal an enlarged, hypokinetic right ventricle with a paper-thin RV free wall. The dilatation of the RV will cause dilatation of the tricuspid valve annulus, with subsequent tricuspid regurgitation. Paradoxical septal motion may also be present.
Cardiac MRI
 MRI in a patient affected by ARVC/D (long axis view of the right ventricle): note the transmural diffuse bright signal in the RV free wall on spin echo T1 (a) due to massive myocardial atrophy with fatty replacement (b).
MRI in a patient affected by ARVC/D (long axis view of the right ventricle): note the transmural diffuse bright signal in the RV free wall on spin echo T1 (a) due to massive myocardial atrophy with fatty replacement (b).
Fatty infiltration of the RV free wall can be visible on cardiac MRI. Fat has increased intensity in T1-weighted images. However, it may be difficult to differentiate intramyocardial fat and the epicardial fat that is commonly seen adjacent to the normal heart. Also, the sub-tricuspid region may be difficult to distinguish from the atrioventricular sulcus, which is rich in fat.
Cardiac MRI can visualize the extreme thinning and akinesis of the RV free wall. However, the normal RV free wall may be about 3 mm thick, making the test less sensitive.
Right ventricular angiography
Right ventricular angiography is considered the gold standard for the diagnosis of ARVD. Findings consistent with ARVD are an akinetic or dyskinetic bulging localized to the infundibular, apical, and subtricuspid regions of the RV. The specificity is 90%; however, the test is observer dependent.
Right ventricular biopsy
Transvenous biopsy of the right ventricle can be highly specific for ARVD, but it has low sensitivity. False positives include other conditions with fatty infiltration of the ventricle, such as chronic alcohol abuse and Duchenne/Becker muscular dystrophy.
False negatives are common, however, because the disease progresses typically from the epicardium to the endocardium (with the biopsy sample coming from the endocardium), and the segmental nature of the disease. Also, due to the paper-thin right ventricular free wall that is common in this disease process, most biopsy samples are taken from the ventricular septum, which is commonly not involved in the disease process.
A biopsy sample that is consistent with ARVD would have > 3% fat, >40% fibrous tissue, and <45% myocytes.
Autopsy
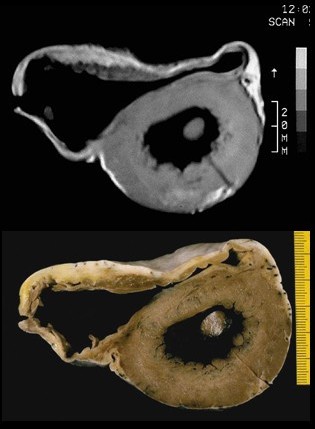
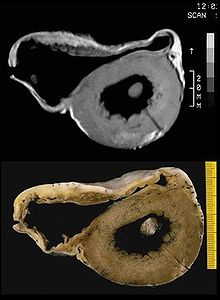 In vitro MRI and corresponding cross section of the heart in ARVD show RV dilatation with anterior and posterior aneurysms (17 year old asymptomatic male athlete who died suddenly during a soccer game).
In vitro MRI and corresponding cross section of the heart in ARVD show RV dilatation with anterior and posterior aneurysms (17 year old asymptomatic male athlete who died suddenly during a soccer game).A post mortem histological demonstration of full thickness substitution of the RV myocardium by fatty or fibro-fatty tissue is consistent with ARVD.
Genetic Testing
ARVD is an autosomal dominant trait with reduced penetrance. Approximately 40-50% of ARVD patients have a mutation identified in one of several genes encoding components of the desmosome, which can help confirm a diagnosis of ARVD.[4] Since ARVD is an autosomal dominant trait, children of an ARVD patient have a 50% chance of inheriting the disease causing mutation. Whenever a mutation is identified by genetic testing, family-specific genetic testing can be used to differentiate between relatives who are at-risk for the disease and those who are not. ARVD genetic testing is clinically available.[5]
Diagnostic Criteria
There is no pathognomonic feature of ARVD. The diagnosis of ARVD is based on a combination of major and minor criteria. To make a diagnosis of ARVD requires either 2 major criteria or 1 major and 2 minor criteria or 4 minor criteria.
Major Criteria
- Right ventricular dysfunction
- Severe dilatation and reduction of RV ejection fraction with little or no LV impairment
- Localized RV aneurysms
- Severe segmental dilatation of the RV
- Tissue characterization
- Fibrofatty replacement of myocardium on endomyocardial biopsy
- Conduction abnormalities
- Epsilon waves in V1 - V3.
- Localized prolongation (>110 ms) of QRS in V1 - V3
- Family history
- Familial disease confirmed on autopsy or surgery
Minor Criteria
- Right ventricular dysfunction
- Mild global RV dilatation and/or reduced ejection fraction with normal LV.
- Mild segmental dilatation of the RV
- Regional RV hypokinesis
- Tissue characterization
- Conduction abnormalities
- Inverted T waves in V2 and V3 in an individual over 12 years old, in the absence of a right bundle branch block (RBBB)
- Late potentials on signal averaged EKG.
- Ventricular tachycardia with a left bundle branch block (LBBB) morphology
- Frequent PVCs (> 1000 PVCs / 24 hours)
- Family history
- Family history of sudden cardiac death before age 35
- Family history of ARVD
Natural history
There is a long asymptomatic lead-time in individuals with ARVD. While this is a genetically transmitted disease, individuals in their teens may not have any characteristics of ARVD on screening tests.
Many individuals have symptoms associated with ventricular tachycardia, such as palpitations, light-headedness, or syncope. Others may have symptoms and signs related to right ventricular failure, such as lower extremity edema, liver congestion with elevated hepatic enzymes. Unfortunately, sudden death may be the first manifestation of disease.
ARVD is a progressive disease. Over time, the right ventricle becomes more involved, leading to right ventricular failure. The right ventricle will fail before there is left ventricular dysfunction. However, by the time the individual has signs of overt right ventricular failure, there will be histological involvement of the left ventricle. Eventually, the left ventricle will also become involved, leading to bi-ventricular failure. Signs and symptoms of left ventricular failure may become evident, including congestive heart failure, atrial fibrillation, and an increased incidence of thromboembolic events.
Management
The goal of management of ARVD is to decrease the incidence of sudden cardiac death. This raises a clinical dilemma: How to prophylactically treat the asymptomatic patient who was diagnosed during family screening.
A certain subgroup of individuals with ARVD are considered at high risk for sudden cardiac death. Characteristics associated with high risk of sudden cardiac death include:
- Young age
- Competitive sports activity
- Malignant familial history
- Extensive RV disease with decreased right ventricular ejection fraction.
- Left ventricular involvement
- Syncope
- Episode of ventricular arrhythmia
Management options include pharmacological, surgical, catheter ablation, and placement of an implantable cardioverter-defibrillator.
Prior to the decision of the treatment option, programmed electrical stimulation in the electrophysiology laboratory may be performed for additional prognostic information. Goals of programmed stimulation include:
- Assessment of the disease's arrhythmogenic potential
- Evaluate the hemodynamic consequences of sustained VT
- Determine whether the VT can be interrupted via antitachycardia pacing.
Regardless of the management option chosen, the individual is typically suggested to undergo lifestyle modification, including avoidance of strenuous exercise, cardiac stimulants (i.e.: caffeine, nicotine, pseudoephedrine) and alcohol. If the individual wishes to begin an exercise regimen, an exercise stress test may have added benefit.
Pharmacologic management
Pharmacologic management of ARVD involves arrhythmia suppression and prevention of thrombus formation.
Sotalol, a beta blocker and a class III antiarrhythmic agent, is the most effective antiarrhythmic agent in ARVD. Other antiarrhythmic agents used include amiodarone and conventional beta blockers (i.e.: metoprolol). If antiarrhythmic agents are used, their efficacy should be guided by series ambulatory holter monitoring, to show a reduction in arrhythmic events.
While angiotensin converting enzyme inhibitors (ACE Inhibitors) are well known for slowing progression in other cardiomyopathies, they have not been proven to be helpful in ARVD.
Individuals with decreased RV ejection fraction with dyskinetic portions of the right ventricle may benefit from long term anticoagulation with warfarin to prevent thrombus formation and subsequent pulmonary embolism.
Catheter ablation
Catheter ablation may be used to treat intractable ventricular tachycardia. It has a 60-90% success rate.[6] Unfortunately, due to the progressive nature of the disease, recurrence is common (60% recurrence rate), with the creation of new arrhythmogenic foci. Indications for catheter ablation include drug-refractory VT and frequent recurrence of VT after ICD placement, causing frequent discharges of the ICD.
Implantable cardioverter-defibrillator
An ICD is the most effective prevention against sudden cardiac death. Due to the prohibitive cost of ICDs, they are not routinely placed in all individuals with ARVD.
Indications for ICD placement in the setting of ARVD include:
- Cardiac arrest due to VT or VF
- Symptomatic VT that is not inducible during programmed stimulation
- Failed programmed stimulation-guided drug therapy
- Severe RV involvement with poor tolerance of VT
- Sudden death of immediate family member
Since ICDs are typically placed via a transvenous approach into the right ventricle, there are complications associated with ICD placement and follow-up.
Due to the extreme thinning of the RV free wall, it is possible to perforate the RV during implantation, potentially causing pericardial tamponade. Because of this, every attempt is made at placing the defibrillator lead on the ventricular septum.
After a successful implantation, the progressive nature of the disease may lead to fibro-fatty replacement of the myocardium at the site of lead placement. This may lead to undersensing of the individual's electrical activity (potentially causing inability to sense VT or VF), and inability to pace the ventricle.
Cardiac transplant surgery
Cardiac transplant surgery may be performed in ARVD. It may be indicated if the arrhythmias associated with the disease are uncontrollable or if there is severe bi-ventricular heart failure that is not manageable with pharmacological therapy.
Family screening
All first degree family members of the affected individual should be screened for ARVD. This is used to establish the pattern of inheritance. Screening should begin during the teenage years unless otherwise indicated. Screening tests include:
- Echocardiogram
- EKG
- Signal averaged EKG
- Holter monitoring
- Cardiac MRI
- Exercise stress test
Highly-publicised deaths from arrhythmogenic right ventricular dysplasia
Sevilla FC and Spanish international left wing-back Antonio Puerta died from the condition, at the age of 22, on 28 August 2007, three days after suffering several cardiac arrests, while disputing a La Liga game against Getafe CF.[7][8] Englishman Matt Gadsby also died from the condition after collapsing on the pitch on 9 September 2006, while playing with his team Hinckley United in a Conference North game against Harrogate Town.[9][10] Model Krissy Taylor, sister of Niki Taylor, died from the disease at age 17 in 1995.
See also
- Woolly hair nevus
References
- ^ Freedberg, et al. (2003). Fitzpatrick's Dermatology in General Medicine. (6th ed.). McGraw-Hill. ISBN 0071380760.
- ^ James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology. (10th ed.). Saunders. ISBN 0721629210.
- ^ Lahtinen, AM; Lehtonen, E, Marjamaa, A, Kaartinen, M, Heliö, T, Porthan, K, Oikarinen, L, Toivonen, L, Swan, H, Jula, A, Peltonen, L, Palotie, A, Salomaa, V, Kontula, K (2011). "Population-prevalent desmosomal mutations predisposing to arrhythmogenic right ventricular cardiomyopathy". Heart rhythm : the official journal of the Heart Rhythm Society 8 (8): 1214–21. doi:10.1016/j.hrthm.2011.03.015. PMID 21397041.
- ^ Sen-Chowdhry S, Syrris P, McKenna WJ (November 2007). "Role of genetic analysis in the management of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy". J. Am. Coll. Cardiol. 50 (19): 1813–21. doi:10.1016/j.jacc.2007.08.008. PMID 17980246.
- ^ Overview of ARVD/C Genetic Testing
- ^ Fontaine G, Tonet J, Gallais Y, Lascault G, Hidden-Lucet F, Aouate P, Halimi F, Poulain F, Johnson N, Charfeddine H, Frank R. (2000). "Ventricular tachycardia catheter ablation in arrhythmogenic right ventricular dysplasia: a 16-year experience". Curr Cardiol Rep 2 (6): 498–506. doi:10.1007/s11886-000-0034-1. PMID 11203287.
- ^ "Sevilla star suffers heart attack". BBC Sport. 2007-08-25. http://news.bbc.co.uk/sport1/hi/football/europe/6964586.stm. Retrieved 2007-08-25.
- ^ Sevilla star dies after collapse
- ^ "Cause of Death". Cardiac Inherited Disease Group. Archived from the original on 2007-09-30. http://web.archive.org/web/20070930080147/http://www.cidg.org/webcontent/default.aspx?tabid=173. Retrieved 2006-10-31.
- ^ "Shock at star player's death". icBirmingham.co.uk. 11 September 2006. http://icbirmingham.icnetwork.co.uk/0100news/0100localnews/tm_objectid=17723437&method=full&siteid=50002&headline=shock-at-star-player-s-death-name_page.html. Retrieved 2007-11-26.
External links
- GeneReviews/NCBI/NIH/UW entry on Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy, Autosomal Dominant
- OMIM entries on Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy, Autosomal Dominant
- http://www.arvd.com
- http://www.arvd.org
- http://www.arvd-arvc-info.com
- http://ourworld.compuserve.com/homepages/drmarknorman/
- http://telethon.bio.unipd.it/ARVDnet/
- http://www.cardiomyopathy.org/html/which_card_arvc.htm
Cytoskeletal defects Microfilaments OtherFibrillin (Marfan syndrome, Weill-Marchesani syndrome, ) · Filamin (FG syndrome 2, Boomerang dysplasia, Larsen syndrome, Terminal osseous dysplasia with pigmentary defects)IF 1/2Keratinopathy (keratosis, keratoderma, hyperkeratosis): KRT1 (Striate palmoplantar keratoderma 3, Epidermolytic hyperkeratosis, IHCM) · KRT2E (Ichthyosis bullosa of Siemens) · KRT3 (Meesmann juvenile epithelial corneal dystrophy) · KRT4 (White sponge nevus) · KRT5 (Epidermolysis bullosa simplex) · KRT8 (Familial cirrhosis) · KRT10 (Epidermolytic hyperkeratosis) · KRT12 (Meesmann juvenile epithelial corneal dystrophy) · KRT13 (White sponge nevus) · KRT14 (Epidermolysis bullosa simplex) · KRT17 (Steatocystoma multiplex) · KRT18 (Familial cirrhosis) · KRT81/KRT83/KRT86 (Monilethrix) · Naegeli–Franceschetti–Jadassohn syndrome · Reticular pigmented anomaly of the flexures345Laminopathy: LMNA (Mandibuloacral dysplasia, Dunnigan Familial partial lipodystrophy, Emery-Dreifuss muscular dystrophy 2, Limb-girdle muscular dystrophy 1B, Charcot–Marie–Tooth disease 2B1) · LMNB (Barraquer–Simons syndrome) · LEMD3 (Buschke–Ollendorff syndrome, Osteopoikilosis) · LBR (Pelger-Huet anomaly, Hydrops-ectopic calcification-moth-eaten skeletal dysplasia)Microtubules OtherTauopathy · Cavernous venous malformationMembrane Catenin Other desmoplakin: Striate palmoplantar keratoderma 2 · Carvajal syndrome · Arrhythmogenic right ventricular dysplasia 8
plectin: Epidermolysis bullosa simplex with muscular dystrophy · Epidermolysis bullosa simplex of Ogna
plakophilin: Skin fragility syndrome · Arrhythmogenic right ventricular dysplasia 9
centrosome: PCNT (Microcephalic osteodysplastic primordial dwarfism type II)Arrestin Myelin Pulmonary surfactant Cell adhesion molecule Tetraspanin TSPAN7 (X-Linked mental retardation 58) · TSPAN12 (Familial exudative vitreoretinopathy 5)Other KIND1 (Kindler syndrome) · HFE (HFE hereditary hemochromatosis) · DYSF (Distal muscular dystrophy, Limb-girdle muscular dystrophy 2B)Genetic disorder, membrane: Channelopathy Calcium channel CACNA1A (Familial hemiplegic migraine 1, Episodic ataxia 2, Spinocerebellar ataxia type-6) · CACNA1C (Timothy syndrome, Brugada syndrome 3, Long QT syndrome 8) · CACNA1F (Ocular albinism 2, CSNB2A) · CACNA1S (Hypokalemic periodic paralysis 1, Thyrotoxic periodic paralysis 1) · CACNB2 (Brugada syndrome 4)Ligand gatedSodium channel Voltage-gatedSCN1A (Familial hemiplegic migraine 3, GEFS+ 2, Febrile seizure 3A) · SCN1B (Brugada syndrome 6, GEFS+ 1) · SCN4A (Hypokalemic periodic paralysis 2, Hyperkalemic periodic paralysis, Paramyotonia congenita, Potassium-aggravated myotonia) · SCN4B (Long QT syndrome 10) · SCN5A (Brugada syndrome 1, Long QT syndrome 3) · SCN9A (Erythromelalgia, Febrile seizure 3B, Paroxysmal extreme pain disorder, Congenital insensitivity to pain)Potassium channel KCNA1 (Episodic ataxia 1) · KCNA5 (Familial atrial fibrillation 7) · KCNC3 (Spinocerebellar ataxia type-13) · KCNE1 (Jervell and Lange-Nielsen syndrome, Long QT syndrome 5) · KCNE2 (Long QT syndrome 6) · KCNE3 (Brugada syndrome 5) · KCNH2 (Short QT syndrome) · KCNQ1 (Jervell and Lange-Nielsen syndrome, Romano-Ward syndrome, Short QT syndrome, Long QT syndrome 1, Familial atrial fibrillation 3) · KCNQ2 (BFNS1}KCNJ1 (Bartter syndrome 2) · KCNJ2 (Andersen-Tawil syndrome, Long QT syndrome 7, Short QT syndrome) · KCNJ11 (TNDM3) · KCNJ18 (Thyrotoxic periodic paralysis 2)Chloride channel TRP channel Connexin GJA1 (Oculodentodigital dysplasia, Hallermann–Streiff syndrome, Hypoplastic left heart syndrome) · GJB1 (Charcot–Marie–Tooth disease X1) · GJB2 (Keratitis–ichthyosis–deafness syndrome, Ichthyosis hystrix, Bart–Pumphrey syndrome, Vohwinkel syndrome) · GJB3/GJB4 (Erythrokeratodermia variabilis, Progressive symmetric erythrokeratodermia) · GJB6 (Clouston's hidrotic ectodermal dysplasia)Porin Categories: - Congenital heart disease

Wikimedia Foundation. 2010.