- X-linked congenital stationary night blindness
-
X-linked congenital stationary night blindness Classification and external resources 
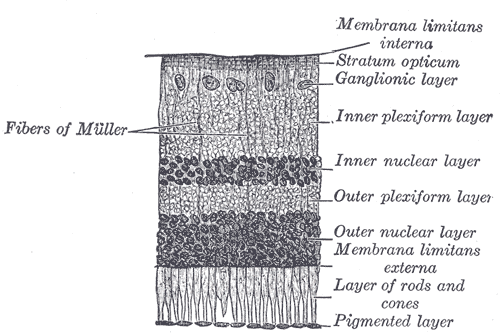
Malfunction in transmission from the photoreceptors in the outer nuclear layer to bipolar cells in the inner nuclear layer underlies CSNB.ICD-10 H53.6 ICD-9 368.61 OMIM 310500 300071 DiseasesDB 32689 MedlinePlus 003039 X-linked congenital stationary night blindness (CSNB) is a rare X-linked non-progressive retinal disorder. It has two forms, complete, also known as type-1 (CSNB1), and incomplete, also known as type-2 (CSNB2), depending on severity. In the complete form (CSNB1), there is no measurable rod cell response to light, whereas this response is measurable in the incomplete form. Patients with this disorder have difficulty adapting to low light situations due to impaired photoreceptor transmission. These patients also often have reduced visual acuity, myopia, nystagmus, and strabismus. CSNB1 is caused by mutations in the gene NYX, which encodes a protein involved in retinal synapse formation or synaptic transmission. CSNB2 is caused by mutations in the gene CACNA1F, which encodes a voltage-gated calcium channel CaV1.4.
Contents
Symptoms
The X-linked varieties of congenital stationary night blindness (CSNB) can be differentiated from the autosomal forms by the presence of myopia, which is typically absent in the autosomal forms. Patients with CSNB often have impaired night vision, myopia, reduced visual acuity, strabismus, and nystagmus. Individuals with the complete form of CSNB (CSNB1) have highly impaired rod sensitivity (reduced ~300x) as well as cone dysfunction. Patients with the incomplete form can present with either myopia or hyperopia.[1]
Cause
CSNB was originally believed to be caused by malfunction in neurotransmission from rods to bipolar cells in the retina. This is due to electroretinogram (ERG) measurements on CSNB patients which show a drastic decrease in the size of the scotopic b-wave in comparison to the a-wave, in CSNB2, or a complete loss of both in CSNB1. The a-wave is believed to represent the response of rods to visual input and remains largely unchanged in CSNB2 patients. The b-wave, however, is believed to result from electrical activity of bipolar cells and is decreased or non-existent in both CSNB1 and 2. CSNB1 patients also show mildly altered cone activity. Further study has demonstrated that the defects found in CSNB patients are better explained by more general defects in both the rod and cone ON-signaling pathways.
Pathophysiology
CSNB1
The complete form of X-linked congenital stationary night blindness, also known as nyctalopia, is caused by mutations in the NYX gene (Nyctalopin on X-chromosome), which encodes a small leucine-rich repeat (LRR) family protein of unknown function.[2][3] This protein consists of an N-terminal signal peptide and 11 LRRs (LRR1-11) flanked by cysteine-rich LRRs (LRRNT and LRRCT). At the C-terminus of the protein there is a putative GPI anchor site. Although the function of NYX is yet to be fully understood, it is believed to be located extracellularly. A naturally occurring deletion of 85 bases in NYX in some mice leads to the "nob" (no b-wave) phenotype, which is highly similar to that seen in CSNB1 patients.[4] NYX is expressed primarily in the rod and cone cells of the retina. There are currently almost 40 known mutations in NYX associated with CSNB1, Table 1., located throughout the protein. As the function of the nyctalopin protein is unknown, these mutations have not been further characterized. However, many of them are predicted to lead to truncated proteins that, presumably, are non-functional.
Table 1. Mutations in NYX associated with CSNB1 Mutation Position References Nucleotide Amino acid c.?-1_?-61del 1_20del Signal sequence [3] Splicing Intron 1 [5] c.?-63_1443-?del 21_481del [3] c.48_64del L18RfsX108 Signal sequence [5] c.85_108del R29_A36del N-terminal LRR [2] c.G91C C31S LRRNT [3] c.C105A C35X LRRNT [3] c.C169A P57T LRRNT [6] c.C191A A64E LRR1 [6] c.G281C R94P LRR2 [7] c.301_303del I101del LRR2 [3] c.T302C I101T LRR2 [7] c.340_351del E114_A118del LRR3 [3], [5] c.G427C A143P LRR4 [3] c.C452T P151L LRR4 [2] c.464_465insAGCGTGCCCGAGCGCCTCCTG S149_V150dup+P151_L155dup LRR4 [2] c.C524G P175R LRR5 [3] c.T551C L184P LRR6 [2] c.556_618delins H186?fsX260 LRR6 [2] c.559_560delinsAA A187K LRR6 [3] c.613_621dup 205_207dup LRR7 [2], [3] c.628_629ins R209_S210insCLR LRR7 [2] c.T638A L213Q LRR7 [2] c.A647G N216S LRR7 [2], [5] c.T695C L232P LRR8 [2] c.727_738del 243_246del LRR8 [3] c.C792G N264K LRR9 [2] c.T854C L285P LRR10 [2] c.T893C F298S LRR10 [2] c.C895T Q299X LRR10 [5] c.T920C L307P LRR11 [3] c.A935G N312S LRR11 [3] c.T1040C L347P LRRCT [3] c.G1049A W350X LRRCT [2] c.G1109T G370V LRRCT [3] c.1122_1457del S374RfsX383 LRRCT [3], [5] c.1306del L437WfsX559 C-terminus [5] LRR: leucine-rich repeat, LRRNT and LRRCT: N- and C-terminal cysteine-rich LRRs. CSNB2
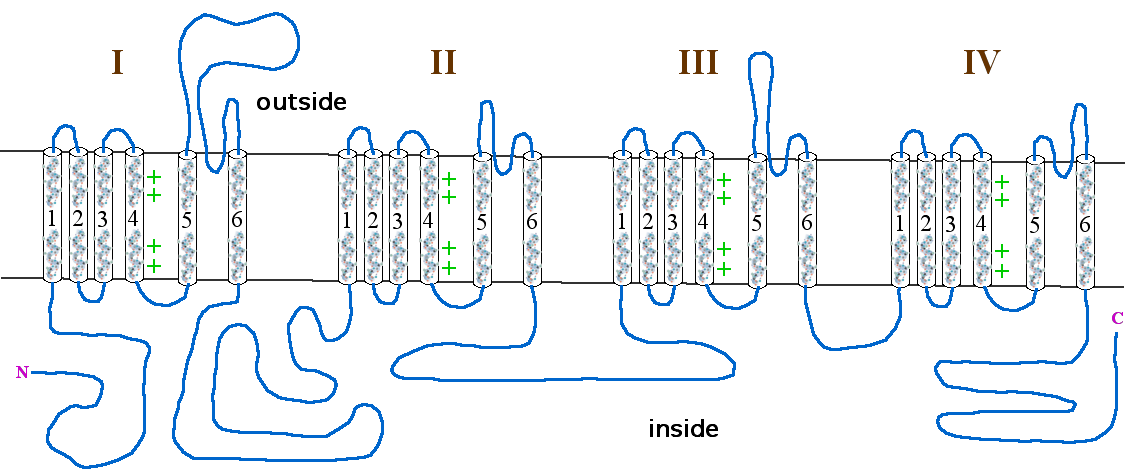
 Figure 1. Schematic structure of CaV1.4 with the domains and subunits labeled.
Figure 1. Schematic structure of CaV1.4 with the domains and subunits labeled.
The incomplete form of X-linked congenital stationary night blindness (CSNB2) is caused by mutations in the CACNA1F gene, which encodes the voltage-gated calcium channel CaV1.4 expressed heavily in retina.[8][9] One of the important properties of this channel is that it inactivates at an extremely low rate. This allows it to produce sustained Ca2+ entry upon depolarization. As photoreceptors depolarize in the absence of light, CaV1.4 channels operate to provide sustained neurotransmitter release upon depolarization.[10] This has been demonstrated in CACNA1F mutant mice that have markedly reduced photoreceptor calcium signals.[11] There are currently 55 mutations in CACNA1F located throughout the channel, Table 2 and Figure 1. While most of these mutations result in truncated and, likely, non-functional channels, it is expected that they prevent the ability of light to hyperpolarize photoreceptors. Of the mutations with known functional consequences, 4 produce channels that are either completely non-functional, and two that result in channels which open at far more hyperpolarized potentials than wild-type. This will result in photoreceptors that continue to release neurotransmitter even after light-induced hyperpolarization.
Table 2. Mutations in CACNA1F associated with CSNB2 Mutation Position Effect References Nucleotide Amino Acid c.C148T R50X N-terminus [12] c.151_155delAGAAA R51PfsX115 N-terminus [13] c.T220C C74R N-terminus [13] c.C244T R82X N-terminus [12], [13] c.466_469delinsGTAGGGGTGCT
CCACCCCGTAGGGGTGCTCCACCS156VdelPinsGVKHOVGVLH D1S2-3 [12], [14], [15] Splicing Intron 4 [12] c.T685C S229P D1S4-5 [13] c.G781A G261R D1-pore [13] c.G832T E278X D1-pore [5], [16] c.904insG R302AfsX314 D1-pore [14] c.951_953delCTT F318del D1-pore [12] c.G1106A G369D D1S6 Activates ~20mV more negative than wild-type, increases time to peak current and decreases inactivation, increased Ca2+ permeability. [8], [10], [12], [13], [17] c.1218delC W407GfsX443 D1-2 [9], [12], [16] c.C1315T Q439X D1-2 [13] c.G1556A R519Q D1-2 Decreased expression [8], [18] c.C1873T R625X D2S4 [12], [13] c.G2021A G674D D2S5 [10], [12], [14] c.C2071T R691X D2-pore [6] c.T2258G F753C D2S6 [13] c.T2267C I756T D2S6 Activates ~35mV more negative than wild-type, inactivates more slowly [19] Splicing Intron 19 [13] c.T2579C L860P D2-3 [13] c.C2683T R895X D3S1-2 [5], [6], [9], [12] Splicing Intron 22 [13], [14] Splicing Intron 22 [13] c.C2783A A928D D3S2-3 [10], [12] c.C2905T R969X D3S4 [8], [13] c.C2914T R972X D3S4 [16] Splicing Intron24 [12] c.C2932T R978X D3S4 [14] c.3006_3008delCAT I1003del D3S4-5 [12] c.G3052A G1018R D3S5 [13] c.3125delG G1042AfsX1076 D3-pore [12] c.3166insC L1056PfsX1066 D3-pore [8], [9], [12], [13] c.C3178T R1060W D3-pore [8], [13] c.T3236C L1079P D3-pore Does not open without BayK, activates ~5mV more negative than wild-type [13], [17] c.3672delC L1225SfsX1266 D4S2 [9], [12] c.3691_3702del G1231_T1234del D4S2 [8], [13] c.G3794T S1265I D4S3 [6] c.C3886A R1296S D4S4 [6] c.C3895T R1299X D4S4 [9], [12], [13] Splicing Intron 32 [13] c.C4075T Q1359X D4-pore [8], [13] c.T4124A L1375H D4-pore Decreased expression [8], [13], [18] Splicing Intron 35 [13] c.G4353A W1451X C-terminus Non-functional [9], [10], [12], [17] c.T4495C C1499R C-terminus [13] c.C4499G P1500R C-terminus [13] c.T4523C L1508P C-terminus [13] Splicing intron 40 [12] c.4581delC F1528LfsX1535 C-terminus [20] c.A4804T K1602X C-terminus [8], [13] c.C5479T R1827X C-terminus [13] c.5663delG S1888TfsX1931 C-terminus [12] c.G5789A R1930H C-terminus [6] Genetics
Only three rhodopsin mutations have been found associated with congenital stationary night blindness (CSNB) [21]. Two of these mutations are found in the second transmembrane helix of rhodopsin at Gly-90 and Thr-94. Specifically, these mutations are the Gly90Asp [22] and the Thr94Ile, which has been the most recent one reported [23]. The third mutation is Ala292Glu, and it is located in the seventh transmembrane helix, in proximity to the site of retinal attachment at Lys-296 [24]. Mutations associated with CSNB affect amino acid residues near the protonated Schiff base (PSB) linkage. They are associated with changes in conformational stability and the protonated status of the PSB nitrogen [25].
External links
Footnotes
- ^ Boycott K, Pearce W, Musarella M, Weleber R, Maybaum T, Birch D, Miyake Y, Young R, Bech-Hansen N (1998). "Evidence for genetic heterogeneity in X-linked congenital stationary night blindness". Am J Hum Genet 62 (4): 865–875. doi:10.1086/301781. PMC 1377021. PMID 9529339. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1377021.
- ^ a b c d e f g h i j k l m n o Bech-Hansen N, Naylor M, Maybaum T, Sparkes R, Koop B, Birch D, Bergen A, Prinsen C, Polomeno R, Gal A, Drack A, Musarella M, Jacobson S, Young R, Weleber R (2000). "Mutations in NYX, encoding the leucine-rich proteoglycan nyctalopin, cause X-linked complete congenital stationary night blindness". Nat Genet 26 (3): 319–323. doi:10.1038/81619. PMID 11062471.
- ^ a b c d e f g h i j k l m n o p q Pusch C, Zeitz C, Brandau O, Pesch K, Achatz H, Feil S, Scharfe C, Maurer J, Jacobi F, Pinckers A, Andreasson S, Hardcastle A, Wissinger B, Berger W, Meindl A (2000). "The complete form of X-linked congenital stationary night blindness is caused by mutations in a gene encoding a leucine-rich repeat protein". Nat Genet 26 (3): 324–327. doi:10.1038/81627. PMID 11062472.
- ^ Gregg R, Mukhopadhyay S, Candille S, Ball S, Pardue M, McCall M, Peachey N (2003). "Identification of the gene and the mutation responsible for the mouse nob phenotype". Invest Ophthalmol Vis Sci 44 (1): 378–384. doi:10.1167/iovs.02-0501. PMID 12506099.
- ^ a b c d e f g h i Zito I, Allen L, Patel R, Meindl A, Bradshaw K, Yates J, Bird A, Erskine L, Cheetham M, Webster A, Poopalasundaram S, Moore A, Trump D, Hardcastle A (2003). "Mutations in the CACNA1F and NYX genes in British CSNBX families". Hum Mutat 21 (2): 169–169. doi:10.1002/humu.9106. PMID 12552565.
- ^ a b c d e f g Zeitz C, Minotti R, Feil S, Mátyás G, Cremers F, Hoyng C, Berger W (2005). "Novel mutations in CACNA1F and NYX in Dutch families with X-linked congenital stationary night blindness". Mol Vis 11: 179–83. PMID 15761389.
- ^ a b Xiao X, Jia X, Guo X, Li S, Yang Z, Zhang Q (2006). "CSNB1 in Chinese families associated with novel mutations in NYX". J Hum Genet 51 (7): 634–640. doi:10.1007/s10038-006-0406-5. PMID 16670814.
- ^ a b c d e f g h i j Strom T, Nyakatura G, Apfelstedt-Sylla E, Hellebrand H, Lorenz B, Weber B, Wutz K, Gutwillinger N, Rüther K, Drescher B, Sauer C, Zrenner E, Meitinger T, Rosenthal A, Meindl A (1998). "An L-type calcium-channel gene mutated in incomplete X-linked congenital stationary night blindness". Nat Genet 19 (3): 260–263. doi:10.1038/940. PMID 9662399.
- ^ a b c d e f g Bech-Hansen N, Naylor M, Maybaum T, Pearce W, Koop B, Fishman G, Mets M, Musarella M, Boycott K (1998). "Loss-of-function mutations in a calcium-channel alpha1-subunit gene in Xp11.23 cause incomplete X-linked congenital stationary night blindness". Nat Genet 19 (3): 264–267. doi:10.1038/947. PMID 9662400.
- ^ a b c d e McRory J, Hamid J, Doering C, Garcia E, Parker R, Hamming K, Chen L, Hildebrand M, Beedle A, Feldcamp L, Zamponi G, Snutch T (2004). "The CACNA1F gene encodes an L-type calcium channel with unique biophysical properties and tissue distribution". J Neurosci 24 (7): 1707–1718. doi:10.1523/JNEUROSCI.4846-03.2004. PMID 14973233.
- ^ Mansergh F, Orton N, Vessey J, Lalonde M, Stell W, Tremblay F, Barnes S, Rancourt D, Bech-Hansen N (2005). "Mutation of the calcium channel gene Cacna1f disrupts calcium signaling, synaptic transmission and cellular organization in mouse retina". Hum Mol Genet 14 (20): 3035–3046. doi:10.1093/hmg/ddi336. PMID 16155113.
- ^ a b c d e f g h i j k l m n o p q r s t Boycott K, Maybaum T, Naylor M, Weleber R, Robitaille J, Miyake Y, Bergen A, Pierpont M, Pearce W, Bech-Hansen N (2001). "A summary of 20 CACNA1F mutations identified in 36 families with incomplete X-linked congenital stationary night blindness, and characterization of splice variants". Hum Genet 108 (2): 91–97. doi:10.1007/s004390100461. PMID 11281458.
- ^ a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac Wutz K, Sauer C, Zrenner E, Lorenz B, Alitalo T, Broghammer M, Hergersberg M, de la Chapelle A, Weber B, Wissinger B, Meindl A, Pusch C (2002). "Thirty distinct CACNA1F mutations in 33 families with incomplete type of XLCSNB and Cacna1f expression profiling in mouse retina". Eur J Hum Genet 10 (8): 449–456. doi:10.1038/sj.ejhg.5200828. PMID 12111638.
- ^ a b c d e Nakamura M, Ito S, Terasaki H, Miyake Y (2001). "Novel CACNA1F mutations in Japanese patients with incomplete congenital stationary night blindness". Invest Ophthalmol Vis Sci 42 (7): 1610–6. PMID 11381068.
- ^ Nakamura M, Ito S, Piao C, Terasaki H, Miyake Y (2003). "Retinal and optic disc atrophy associated with a CACNA1F mutation in a Japanese family". Arch Ophthalmol 121 (7): 1028–1033. doi:10.1001/archopht.121.7.1028. PMID 12860808.
- ^ a b c Allen L, Zito I, Bradshaw K, Patel R, Bird A, Fitzke F, Yates J, Trump D, Hardcastle A, Moore A (2003). "Genotype-phenotype correlation in British families with X linked congenital stationary night blindness". Br J Ophthalmol 87 (11): 1413–1420. doi:10.1136/bjo.87.11.1413. PMC 1771890. PMID 14609846. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1771890.
- ^ a b c Hoda J, Zaghetto F, Koschak A, Striessnig J (2005). "Congenital stationary night blindness type 2 mutations S229P, G369D, L1068P, and W1440X alter channel gating or functional expression of Ca(v)1.4 L-type Ca2+ channels". J Neurosci 25 (1): 252–259. doi:10.1523/JNEUROSCI.3054-04.2005. PMID 15634789.
- ^ a b Hoda J, Zaghetto F, Singh A, Koschak A, Striessnig J (2006). "Effects of congenital stationary night blindness type 2 mutations R508Q and L1364H on Cav1.4 L-type Ca2+ channel function and expression". J Neurochem 96 (6): 1648–1658. doi:10.1111/j.1471-4159.2006.03678.x. PMID 16476079.
- ^ Hemara-Wahanui A, Berjukow S, Hope C, Dearden P, Wu S, Wilson-Wheeler J, Sharp D, Lundon-Treweek P, Clover G, Hoda J, Striessnig J, Marksteiner R, Hering S, Maw M (2005). "A CACNA1F mutation identified in an X-linked retinal disorder shifts the voltage dependence of Cav1.4 channel activation". Proc Natl Acad Sci USA 102 (21): 7553–7558. doi:10.1073/pnas.0501907102. PMC 1140436. PMID 15897456. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1140436.
- ^ Jacobi F, Hamel C, Arnaud B, Blin N, Broghammer M, Jacobi P, Apfelstedt-Sylla E, Pusch C (2003). "A novel CACNA1F mutation in a french family with the incomplete type of X-linked congenital stationary night blindness". Am J Ophthalmol 135 (5): 733–736. doi:10.1016/S0002-9394(02)02109-8. PMID 12719097.
- ^ Pere Garriga, and Joan Manyosa. The eye photoreceptor protein rhodopsin. Structural implications for retinal disease. Volume 528, Issues 1-3, 25 September 2002, Pages 17- 22.
- ^ V.R. Rao, G.B. Cohen and D.D. Oprian Nature 367 (1994), pp. 639–642.
- ^ N. al-Jandal, G.J. Farrar, A.S. Kiang, M.M. Humphries, N. Bannon, J.B. Findlay, P. Humphries and P.F. Kenna Hum. Mutat. 13 (1999), pp. 75–81.
- ^ T.P. Dryja, E.L. Berson, V.R. Rao and D.D. Oprian Nat. Genet. 4 (1993), pp. 280–283.
- ^ P.A. Sieving, J.E. Richards, F. Naarendorp, E.L. Bingham, K. Scott and M. Alpern Proc. Natl. Acad. Sci. USA 92 (1995), pp. 880–884.
Categories:- Channelopathy
- Diseases of the eye and adnexa
Wikimedia Foundation. 2010.