- Generalized epilepsy with febrile seizures plus
-
Generalized epilepsy with febrile seizures plus (GEFS+) is a syndromic autosomal dominant disorder where afflicted individuals can exhibit numerous epilepsy phenotypes.[1] GEFS+ can persist beyond early childhood (i.e., 6 years of age). GEFS+ is also now believed to encompass three other epilepsy disorders: severe myoclonic epilepsy of infancy (SMEI), which is also known as Dravet's syndrome, borderline SMEI (SMEB), and intractable epilepsy of childhood (IEC).[2][3] There are at least five types of GEFS+, delineated by their causative gene. Known causative genes are the sodium channel α subunit genes SCN1A and SCN2A, an associated β subunit SCN1B, and a GABAA receptor γ subunit gene, GABRG2. Penetrance for this disorder is estimated at approximately 60%.[4]
GEFS+ Classification and external resources ICD-10 G40.3 OMIM 604233 609800 607208 Contents
Symptoms and signs
Individuals with GEFS+ present with a range of epilepsy phenotypes. These include febrile seizures that end by age 6 (FS), such seizures extending beyond age 6 that may include afebrile tonic-clonic, myoclonic, absence, atonic seizures and myoclonic-astatic epilepsy. Individuals may also present with SMEI, which is characterized by generally tonic-clonic seizures, impaired psychomotor development, myoclonic seizures, ataxia, and poor response to many epileptic drugs.[1][5]
Diagnosis
Pathophysiology
Type 1
 Figure 1. Schematic structure of SCN1B with GEFS+ type 1 mutations shown in red. The single red spot is the C121W mutant at the disulfide bond (black) and the stretch of red the I70_E74del mutation.
Figure 1. Schematic structure of SCN1B with GEFS+ type 1 mutations shown in red. The single red spot is the C121W mutant at the disulfide bond (black) and the stretch of red the I70_E74del mutation.
GEFS+ type 1 is a subtype of GEFS+ in which there are mutations in SCN1B, a gene encoding a sodium channel β subunit. The β subunit is required for proper channel inactivation. There are two known mutations in SCN1B that lead to GEFS+ (Figure 1). The first and best characterized of these mutations is C121W. This mutation alters a cysteine involved in a disulfide bond in the extracellular N-terminus of the protein. This extracellular region is similar to the cell adhesion molecule contactin and other cell adhesion molecules. It is believed that the disulfide bond disrupted by the C121W mutation is required for the proper folding of this N-terminus motif. Coexpression of SCN1B with sodium channel α subunits in oocytes and other cells results in channels that inactivate more slowly. Expression of C121W mutant along with wild-type α subunits produces current indistinguishable from that through α subunits alone.[4][6] Further investigation of this mutation has indicated that it results in decreased frequency dependent rundown and, thus, likely hyperexcitability when compared to cells expressing the wild-type subunit. Interestingly, this mutation also disrupts the subunit's ability to induce cellular aggregation. The importance of this last fact is unclear, though it is presumed that proper channel aggregation within cells and cell-cell contact are required for normal neuronal function.[7][8]
A second mutation has been found in one kindred with GEFS+ type 1. This mutation is in a splice acceptor site of exon 3. The loss of this acceptor site reveals a downstream cryptic acceptor site and a protein missing 5 amino acids in the N-terminus (I70_E74del). This mutation has not been further characterized.[9]
Type 2
A second subtype of GEFS+, type 2, is the result of mutations in SCN1A, a gene encoding a sodium channel α subunit. There are currently almost 90 known mutations in the SCN1A gene throughout the entirety of the channel (see table 1). These mutations result in almost any imaginable mutation type in the gene, short of duplications. The results of these mutations are highly variable, some producing functional channels while others result in non-functional channels. Some functional channels result in membrane hyperexcitability while others result in hypoexcitability. Most of the functional mutant channels result in hyperexcitability due to decreased frequency dependent rundown. An example of this is the D188V mutation. A 10 Hz stimulation of wild-type channels causes current to decrease to approximately 70% of maximum whereas the same stimulation of mutant channels results in rundown to 90% of maximum. This is causes by an expedited recovery from inactivation for mutant channels versus wild-type. The D188V mutant, for example, recovers to 90% maximal current in 200ms while wild-type channels are unable to recover to this degree in >1000ms.[10] Some other functional mutations that lead to hyperexcitability due so by other means, such as decreasing the rate of entrance into the slow inactivated state.
Some of the other functional mutations are believed to result in hypoexcitability. The R859C mutation, for example, has a more depolarized voltage dependence of activation, meaning that the membrane must be more depolarized for the channel to open. This mutant also recovers more slowly from inactivation.[11] The nonfunctional channels are believed to produce similar changes in cell excitability. Likewise, many of the nonsense mutations likely result in nonfunctional channels and hypoexcitability, though this has yet to be tested. It is also unclear how this membrane hypoexcitability leads to the GEFS+ phenotype.
Table 1. Summary of mutations found in patients diagnosed with GEFS+ type 2 Mutation Region Functional? Excitability Prediction References R101Q N-Terminus [12] S103G N-Terminus [13] T112I N-Terminus [13] V144fsX148 D1S1 [12] G177fsX180 D1S2-S3 [13] D188V D1S2-S3 Yes Hyperexcitable [10][14] F190R D1S3 [12] S219fsX275 D1S4 [15] R222X D1S4 [12][15] G265W D1S5 [13] G343E D1S5-S6 [13] E435X D1-2 [12] R613X D1-2 [16] R701X D1-2 [12] P707fsX715 D1-2 [16] R712X D1-2 [13] Q732fsX749 D1-2 [13] Y779C D2S1 [17] T808S D2S2 Yes Hyperexcitable [5][13] R859C D2S4 Yes Hypoexcitability [11] T875M D2S4 Yes Hyperexcitable* [18][19][20][21][22] F902C D2S5 No Hypoexcitable [23] S914fsX934 D2S5-6 [16] M924I D2S5-6 [12] V934A D2S5-6 [12] R936C D2S5-6 [12] R936H D2S5-6 [12] W942X D2S5-6 [12] R946fsX953 D2S5-6 [13] W952X D2S5-6 [13] D958fsX973 D2S5-6 [13] M960V D2S5-6 [13] G979R D2S6 No Hypoexcitable [5][13] V983A D2S6 Yes Hyperexcitable [5][13] N985I D2S6 [13] L986F D2S6 No Hypoexcitable [15][24] N1011I D2-3 Yes Hyperexcitable [5][13] K1100fsX1107 D2-3 [15] L1156fsX1172 D2-3 [12] W1204R D2-3 Yes Hyperexcitable [2][22][25] W1204X D2-3 [13] R1213X D2-3 [13] S1231R D3S1 [13] S1231T D3S1 [16] F1263L D3S2 [13] W1284X D3S3 [13] L1345P D3S5 [12] V1353L D3S5 No Hypoexcitable [14][24] Splice Exon 4 [13][15] R1397X D3S5-6 [12] R1407X D3S5-6 [13] W1408X D3S5-6 [13] V1428A D3S6 [26][27] S1516X D3-4 [13] R1525X D3-4 [16] M1549del D4S1 [12] V1611F D4S3 Yes Hyperexcitable [5][13] P1632S D4S3 Yes Hyperexcitable [5][13] R1635X D4S4 [12] R1648C D4S4 Yes Hyperexcitable [23] R1648H D4S4 Yes Hyperexcitable [19][21][22][28][29] I1656M D4S4 Yes [14][24] R1657C D4S4 Yes Hypoexcitable [24][29][30] F1661S D4S4 Yes Hyperexcitable [23] L1670fsX1678 D4S4-5 [13][15] G1674R D4S4-5 No Hypoexcitable [23] F1682S D4S5 [12] Y1684C D4S5 [12] A1685V D4S5 No Hypoexcitable [24][26][27] A1685D D4S5 [13] T1709I D4S5-6 No Hypoexcitable [5][13] D1742G D4S5-6 [31] G1749E D4S6 Yes Hypoexcitable [23] F1756del D4S6 [12] F1765fsX1794 D4S6 [13] Y1771C D4S6 [12] 1807delMFYE C-Terminus [13] F1808L C-Terminus Yes Hyperexcitable [5][13] W1812G C-Terminus [13] F1831S C-Terminus [13] M1841T C-Terminus [17] S1846fsX1856 C-Terminus [15][16] R1882X C-Terminus [12] D1886Y C-Terminus Yes Hyperexcitable [32] R1892X C-Terminus [13] R1902X C-Terminus [12] Q1904fsX1945 C-Terminus [13] *Results are dependent on experimental paradigm Type 3
 Figure 2. Schematic structure of GABRG2 with the GEFS+ type 3 mutations noted in red.
Figure 2. Schematic structure of GABRG2 with the GEFS+ type 3 mutations noted in red.Patients with GEFS+ type 3 have mutations in the GABRG2 gene, which encodes the GABAA γ2 subunit (figure 2). The first mutation discovered in GABRG2 was K289M, in the extracellular region linking membrane-spanning domains M2 and M3. Oocytes injected with α1, β2, and γ2 subunits produce large GABA inducible currents whereas those injected with K289M mutant instead of wild-type subunits produce currents much smaller (about 10% of wild-type). This abnormal current is not the result of non-incorporation of mutant subunits since mutant containing receptors are still sensitive to benzodiazepines, a property for which functional γ subunits are required. Because of these results, it is believed that the GEFS+ phenotype in these individuals is a result of hyperexcitability.[33]
Concurrent with the previous mutation, a second group found a second mutation in GABRG2 associated with GEFS+. This mutation, R43Q, is located in the one of two benzodiazepine binding-sites located in the extracellular N-terminus. Benzodiazepines, such as Diazepam, potentiate GABA induced current. This potentiation is abolished in cells expressing the R43Q mutant subunit instead of the wild-type γ subunit. This mutation does not affect the subunit's ability to coassemble into function receptors as it still confers resistance to GABA current blockade by zinc. As with the previous mutation, this mutation is expected to result in neuronal hyperexcitability.[34][35]
The final known GEFS+ type 3 mutation is a nonsense mutation, Q351X, located in the intracellular region linking the third and fourth membrane spanning segments. When this mutant subunit is expressed in cells with wild-type α and β subunits it produces non-functional receptors. Since wild-type α and β subunits expressed alone are able to produce GABA inducible current this indicates that the mutation either prevents both coassembly of the mutant and wild-type subunits but also coassembly of the wild-type α and β subunits or prevents proper trafficking of the formed receptor to the membrane. Fusion of GFP onto this mutated subunit has indicated that it is localized to the endoplasmic reticulum instead of the cell membrane. As with other known GEFS+ type 3 mutation, Q351X likely results in neuronal hyperexcitability.[36]
SCN2A mutations
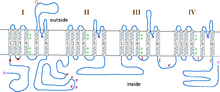 Figure 3. Schematic structure of SCN2A with GEFS+ associated mutation positions indicated by red dots.
Figure 3. Schematic structure of SCN2A with GEFS+ associated mutation positions indicated by red dots.The final type of GEFS+ is caused by mutations in the SCN2A gene, which encodes a sodium channel α subunit. The first associated mutation in this gene is R187W, located on the intracellular region linking membrane spanning units two and three in the first domain (D1S2-S3, figure 3). Patients with this mutation have both febrile and afebrile seizures. Electrophysiological examination of this mutant revealed that it increases the time constant for inactivation, presumably increasing sodium current and leading to hyperexcitability. However, this mutation also yields channels that inactivate at more hyperpolarized potentials relative to wild-type channels, indicative of hypoexcitability. Whether the end result on membrane excitability of this mutation is hyperexcitability or hypoexcitability is, as yet, unclear.[27][37]
The second known mutation in SCN2A associated with GEFS+ is R102X. This mutation is located in the intracellular N-terminus (figure 3) and results in SMEI in patients. The result of this mutation is completely non-functional channels and membrane hypoexcitability. Interestingly, the truncated mutant protein also seems to cause wild-type channels to inactivate at more hyperpolarized potentials, indicating that it also acts in a dominant negative manner.[38]
Treatment/Management
Children and Adults with Dravet syndrome experience multiple seizure types that are resistant to most anti-epileptic medications. Currently, the evidence supports the use of “rational polytherapy” which consists of a step-wise introduction of medications that have been shown to improve seizure control in patients with Dravet syndrome until the patient either responds favorably or experiences unacceptable side effects. It must be emphasized that significant differences exist between countries with regard to drug dose preferences and availability of anti-epileptic medications.
The following medications have been shown to benefit patients with Dravet syndrome[39]:
- divalproex sodium and derivatives (Depakote, Depakene,Epilim, Epival, Micropakine)
- topiramate (Topamax)
- stiripentol (Diacomit)
- clobazam (Frisium, Urbanyl)
- clonazepam (Klonipin, Rivotril)
- levetiracetam (Keppra)
- bromides
The following medications may aggravate seizures in Dravet syndrome[39][40]:
- lamotrigine (Lamictal)
- phenytoin (Dilantin, Epanutin)
- fosphenytoin (Cerebyx, Prodilantin)
- carbamazepine (Tegretol, Calepsin, Cargagen, Barbatrol, Epitol, Finlepsin, Sirtal, Stazepine)
- oxcarbazepine (Trileptal)
- vigabatrin (Sabril, Sabrilan, Sabrilex)
Non-pharmacologic therapy with the ketogenic diet has been shown to improve seizure control in a significant percentage of children with Dravet syndrome.[41]
Focal resective surgery is usually not helpful as SMEI is a systemic disorder without identifiable focal pathology.
Epidemiology
Febrile seizures affect approximately 6% of the population.
See also
- Febrile seizures
- Idiopathic generalized epilepsy
- Dravet Syndrome Foundation
- International Dravet Epilepsy Action League
External links
Advocacy Organizations:
- Dravet Italia Onlus
- Dravet Syndrome Foundation
- Dravet Syndrome Foundation Spain (DSF Spain)
- Dravet Syndrome UK
- ICE Alliance
- IDEA League
References
- ^ a b Scheffer I, Berkovic S (1997). "Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes". Brain 120 (3) (3): 479–90. doi:10.1093/brain/120.3.479. PMID 9126059.
- ^ a b Spampanato J, Escayg A, Meisler M, Goldin A (2003). "Generalized epilepsy with febrile seizures plus type 2 mutation W1204R alters voltage-dependent gating of Na(v)1.1 sodium channels". Neuroscience 116 (1): 37–48. doi:10.1016/S0306-4522(02)00698-X. PMID 12535936.
- ^ Singh R, Andermann E, Whitehouse W, Harvey A, Keene D, Seni M, Crossland K, Andermann F, Berkovic S, Scheffer I (2001). "Severe myoclonic epilepsy of infancy: extended spectrum of GEFS+?". Epilepsia 42 (7): 837–44. doi:10.1046/j.1528-1157.2001.042007837.x. PMID 11488881.
- ^ a b Wallace R, Wang D, Singh R, Scheffer I, George A, Phillips H, Saar K, Reis A, Johnson E, Sutherland G, Berkovic S, Mulley J (1998). "Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B". Nat Genet 19 (4): 366–70. doi:10.1038/1252. PMID 9697698.
- ^ a b c d e f g h i Rhodes T, Vanoye C, Ohmori I, Ogiwara I, Yamakawa K, George A (2005). "Sodium channel dysfunction in intractable childhood epilepsy with generalized tonic–clonic seizures". J Physiol 569 (Pt 2): 433–45. doi:10.1113/jphysiol.2005.094326. PMC 1464244. PMID 16210358. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1464244.
- ^ Tammaro P, Conti F, Moran O (2002). "Modulation of sodium current in mammalian cells by an epilepsy-correlated beta 1-subunit mutation". Biochem Biophys Res Commun 291 (4): 1095–101. doi:10.1006/bbrc.2002.6570. PMID 11866477.
- ^ Meadows L, Malhotra J, Loukas A, Thyagarajan V, Kazen-Gillespie K, Koopman M, Kriegler S, Isom L, Ragsdale D (2002). "Functional and biochemical analysis of a sodium channel beta1 subunit mutation responsible for generalized epilepsy with febrile seizures plus type 1". J Neurosci 22 (24): 10699–709. PMID 12486163.
- ^ Lucas P, Meadows L, Nicholls J, Ragsdale D (2005). "An epilepsy mutation in the beta1 subunit of the voltage-gated sodium channel results in reduced channel sensitivity to phenytoin". Epilepsy Res 64 (3): 77–84. doi:10.1016/j.eplepsyres.2005.03.003. PMID 15922564.
- ^ Audenaert D, Claes L, Ceulemans B, Löfgren A, Van Broeckhoven C, De Jonghe P (2003). "A deletion in SCN1B is associated with febrile seizures and early-onset absence epilepsy". Neurology 61 (6): 854–6. PMID 14504340.
- ^ a b Cossette P, Loukas A, Lafrenière R, Rochefort D, Harvey-Girard E, Ragsdale D, Dunn R, Rouleau G (2003). "Functional characterization of the D188V mutation in neuronal voltage-gated sodium channel causing generalized epilepsy with febrile seizures plus (GEFS)". Epilepsy Res 53 (1–2): 107–17. doi:10.1016/S0920-1211(02)00259-0. PMID 12576172.
- ^ a b Barela A, Waddy S, Lickfett J, Hunter J, Anido A, Helmers S, Goldin A, Escayg A (2006). "An epilepsy mutation in the sodium channel SCN1A that decreases channel excitability". J Neurosci 26 (10): 2714–23. doi:10.1523/JNEUROSCI.2977-05.2006. PMID 16525050.
- ^ a b c d e f g h i j k l m n o p q r s t u v Fukuma G, Oguni H, Shirasaka Y, Watanabe K, Miyajima T, Yasumoto S, Ohfu M, Inoue T, Watanachai A, Kira R, Matsuo M, Muranaka H, Sofue F, Zhang B, Kaneko S, Mitsudome A, Hirose S (2004). "Mutations of neuronal voltage-gated Na+ channel alpha 1 subunit gene SCN1A in core severe myoclonic epilepsy in infancy (SMEI) and in borderline SMEI (SMEB)". Epilepsia 45 (2): 140–8. doi:10.1111/j.0013-9580.2004.15103.x. PMID 14738421.
- ^ a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae af ag ah ai aj ak Fujiwara T, Sugawara T, Mazaki-Miyazaki E, Takahashi Y, Fukushima K, Watanabe M, Hara K, Morikawa T, Yagi K, Yamakawa K, Inoue Y (2003). "Mutations of sodium channel alpha subunit type 1 (SCN1A) in intractable childhood epilepsies with frequent generalized tonic-clonic seizures". Brain 126 (Pt 3): 531–46. doi:10.1093/brain/awg053. PMID 12566275.
- ^ a b c Wallace R, Scheffer I, Barnett S, Richards M, Dibbens L, Desai R, Lerman-Sagie T, Lev D, Mazarib A, Brand N, Ben-Zeev B, Goikhman I, Singh R, Kremmidiotis G, Gardner A, Sutherland G, George A, Mulley J, Berkovic S (2001). "Neuronal Sodium-Channel α1-Subunit Mutations in Generalized Epilepsy with Febrile Seizures Plus". Am J Hum Genet 68 (4): 859–65. doi:10.1086/319516. PMC 1275639. PMID 11254444. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1275639.
- ^ a b c d e f g Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P (2001). "De Novo Mutations in the Sodium-Channel Gene SCN1A Cause Severe Myoclonic Epilepsy of Infancy". Am J Hum Genet 68 (6): 1327–32. doi:10.1086/320609. PMC 1226119. PMID 11359211. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1226119.
- ^ a b c d e f Kearney J, Wiste A, Stephani U, Trudeau M, Siegel A, RamachandranNair R, Elterman R, Muhle H, Reinsdorf J, Shields W, Meisler M, Escayg A (2006). "Recurrent de novo mutations of SCN1A in severe myoclonic epilepsy of infancy". Pediatr Neurol 34 (2): 116–20. doi:10.1016/j.pediatrneurol.2005.07.009. PMID 16458823.
- ^ a b Annesi G, Gambardella A, Carrideo S, Incorpora G, Labate A, Pasqua A, Civitelli D, Polizzi A, Annesi F, Spadafora P, Tarantino P, Cirò Candiano I, Romeo N, De Marco E, Ventura P, LePiane E, Zappia M, Aguglia U, Pavone L, Quattrone A (2003). "Two novel SCN1A missense mutations in generalized epilepsy with febrile seizures plus". Epilepsia 44 (9): 1257–8. doi:10.1046/j.1528-1157.2003.22503.x. PMID 12919402.
- ^ Moulard B, Guipponi M, Chaigne D, Mouthon D, Buresi C, Malafosse A (1999). "Identification of a New Locus for Generalized Epilepsy with Febrile Seizures Plus (GEFS+) on Chromosome 2q24-q33". Am J Hum Genet 65 (5): 1396–400. doi:10.1086/302621. PMC 1288292. PMID 10521305. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1288292.
- ^ a b Escayg A, MacDonald B, Meisler M, Baulac S, Huberfeld G, An-Gourfinkel I, Brice A, LeGuern E, Moulard B, Chaigne D, Buresi C, Malafosse A (2000). "Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2". Nat Genet 24 (4): 343–5. doi:10.1038/74159. PMID 10742094.
- ^ Alekov A, Rahman M, Mitrovic N, Lehmann-Horn F, Lerche H (2001). "Enhanced inactivation and acceleration of activation of the sodium channel associated with epilepsy in man". Eur J Neurosci 13 (11): 2171–6. doi:10.1046/j.0953-816x.2001.01590.x. PMID 11422459.
- ^ a b Spampanato J, Escayg A, Meisler M, Goldin A (2001). "Functional effects of two voltage-gated sodium channel mutations that cause generalized epilepsy with febrile seizures plus type 2". J Neurosci 21 (19): 7481–90. PMID 11567038.
- ^ a b c Lossin C, Wang D, Rhodes T, Vanoye C, George A (2002). "Molecular basis of an inherited epilepsy". Neuron 34 (6): 877–84. doi:10.1016/S0896-6273(02)00714-6. PMID 12086636.
- ^ a b c d e Rhodes T, Lossin C, Vanoye C, Wang D, George A (2004). "Noninactivating voltage-gated sodium channels in severe myoclonic epilepsy of infancy". Proc Natl Acad Sci USA 101 (30): 11147–52. doi:10.1073/pnas.0402482101. PMC 503754. PMID 15263074. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=503754.
- ^ a b c d e Lossin C, Rhodes T, Desai R, Vanoye C, Wang D, Carniciu S, Devinsky O, George A (2003). "Epilepsy-associated dysfunction in the voltage-gated neuronal sodium channel SCN1A". J Neurosci 23 (36): 11289–95. PMID 14672992.
- ^ Escayg A, Heils A, MacDonald B, Haug K, Sander T, Meisler M (2001). "A Novel SCN1A Mutation Associated with Generalized Epilepsy with Febrile Seizures Plus—and Prevalence of Variants in Patients with Epilepsy". Am J Hum Genet 68 (4): 866–73. doi:10.1086/319524. PMC 1275640. PMID 11254445. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1275640.
- ^ a b Ito M, Nagafuji H, Okazawa H, Yamakawa K, Sugawara T, Mazaki-Miyazaki E, Hirose S, Fukuma G, Mitsudome A, Wada K, Kaneko S (2002). "Autosomal dominant epilepsy with febrile seizures plus with missense mutations of the (Na+)-channel alpha 1 subunit gene, SCN1A". Epilepsy Res 48 (1–2): 15–23. doi:10.1016/S0920-1211(01)00313-8. PMID 11823106.
- ^ a b c Ito M, Yamakawa K, Sugawara T, Hirose S, Fukuma G, Kaneko S (2006). "Phenotypes and genotypes in epilepsy with febrile seizures plus". Epilepsy Res 70 (2–3 Suppl): 199–205. doi:10.1016/j.eplepsyres.2005.11.028. PMID 16884893.
- ^ Baulac S, Gourfinkel-An I, Picard F, Rosenberg-Bourgin M, Prud'homme J, Baulac M, Brice A, LeGuern E (1999). "A Second Locus for Familial Generalized Epilepsy with Febrile Seizures Plus Maps to Chromosome 2q21-q33". Am J Hum Genet 65 (4): 1078–85. doi:10.1086/302593. PMC 1288241. PMID 10486327. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1288241.
- ^ a b Vanoye C, Lossin C, Rhodes T, George A (2006). "Single-channel Properties of Human NaV1.1 and Mechanism of Channel Dysfunction in SCN1A-associated Epilepsy". J Gen Physiol 127 (1): 1–14. doi:10.1085/jgp.200509373. PMC 2151481. PMID 16380441. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2151481.
- ^ Nagao Y, Mazaki-Miyazaki E, Okamura N, Takagi M, Igarashi T, Yamakawa K (2005). "A family of generalized epilepsy with febrile seizures plus type 2-a new missense mutation of SCN1A found in the pedigree of several patients with complex febrile seizures". Epilepsy Res 63 (2–3): 151–6. doi:10.1016/j.eplepsyres.2004.11.005. PMID 15715999.
- ^ Pineda-Trujillo N, Carrizosa J, Cornejo W, Arias W, Franco C, Cabrera D, Bedoya G, Ruíz-Linares A (2005). "A novel SCN1A mutation associated with severe GEFS+ in a large South American pedigree". Seizure 14 (2): 123–8. doi:10.1016/j.seizure.2004.12.007. PMID 15694566.
- ^ Spampanato J, Kearney J, de Haan G, McEwen D, Escayg A, Aradi I, MacDonald B, Levin S, Soltesz I, Benna P, Montalenti E, Isom L, Goldin A, Meisler M (2004). "A novel epilepsy mutation in the sodium channel SCN1A identifies a cytoplasmic domain for beta subunit interaction". J Neurosci 24 (44): 10022–34. doi:10.1523/JNEUROSCI.2034-04.2004. PMID 15525788.
- ^ Baulac S, Huberfeld G, Gourfinkel-An I, Mitropoulou G, Beranger A, Prud'homme J, Baulac M, Brice A, Bruzzone R, LeGuern E (2001). "First genetic evidence of GABA(A) receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene". Nat Genet 28 (1): 46–8. doi:10.1038/88254. PMID 11326274.
- ^ Wallace R, Marini C, Petrou S, Harkin L, Bowser D, Panchal R, Williams D, Sutherland G, Mulley J, Scheffer I, Berkovic S (2001). "Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures". Nat Genet 28 (1): 49–52. doi:10.1038/88259. PMID 11326275.
- ^ Marini C, Harkin L, Wallace R, Mulley J, Scheffer I, Berkovic S (2003). "Childhood absence epilepsy and febrile seizures: a family with a GABA(A) receptor mutation". Brain 126 (Pt 1): 230–40. doi:10.1093/brain/awg018. PMID 12477709.
- ^ Harkin L, Bowser D, Dibbens L, Singh R, Phillips F, Wallace R, Richards M, Williams D, Mulley J, Berkovic S, Scheffer I, Petrou S (2002). "Truncation of the GABAA-Receptor γ2 Subunit in a Family with Generalized Epilepsy with Febrile Seizures Plus". Am J Hum Genet 70 (2): 530–6. doi:10.1086/338710. PMC 384926. PMID 11748509. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=384926.
- ^ Sugawara T, Tsurubuchi Y, Agarwala K, Ito M, Fukuma G, Mazaki-Miyazaki E, Nagafuji H, Noda M, Imoto K, Wada K, Mitsudome A, Kaneko S, Montal M, Nagata K, Hirose S, Yamakawa K (2001). "A missense mutation of the Na+ channel αII subunit gene Nav1.2 in a patient with febrile and afebrile seizures causes channel dysfunction". Proc Natl Acad Sci USA 98 (11): 6384–9. doi:10.1073/pnas.111065098. PMC 33477. PMID 11371648. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=33477.
- ^ Kamiya K, Kaneda M, Sugawara T, Mazaki E, Okamura N, Montal M, Makita N, Tanaka M, Fukushima K, Fujiwara T, Inoue Y, Yamakawa K (2004). "A nonsense mutation of the sodium channel gene SCN2A in a patient with intractable epilepsy and mental decline". J Neurosci 24 (11): 2690–8. doi:10.1523/JNEUROSCI.3089-03.2004. PMID 15028761.
- ^ a b http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=gefs
- ^ http://idea-league.org/care-and-treatment
- ^ http://onlinelibrary.wiley.com/doi/10.1111/j.1528-1167.2005.05705.x/abstract
Categories:- Channelopathy
- Epilepsy types
Wikimedia Foundation. 2010.