- Familial adenomatous polyposis
-
Familial adenomatous polyposis Classification and external resources 
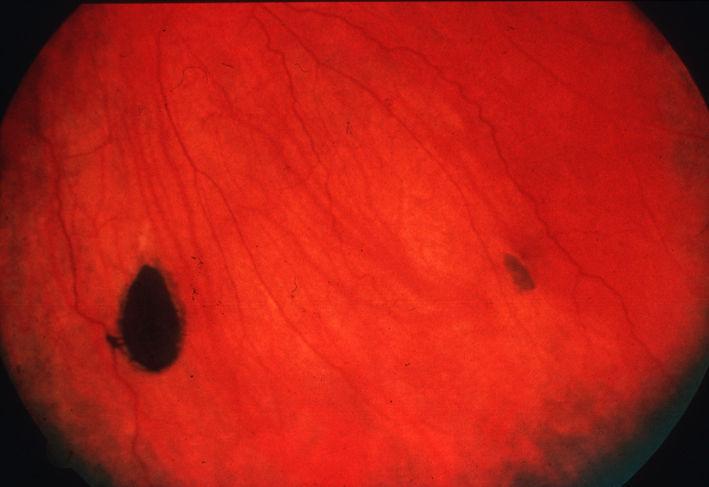
Endoscopic image of sigmoid colon of patient with familial adenomatous polyposis.ICD-10 C18, D12 ICD-9 211.3 ICD-O: 8220/0 OMIM 175100 DiseasesDB 4678 eMedicine med/769 MeSH D011125 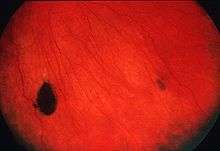 CHRPE
CHRPE
Familial adenomatous polyposis (FAP) is an inherited condition in which numerous polyps form mainly in the epithelium of the large intestine. While these polyps start out benign, malignant transformation into colon cancer occurs when not treated.
Contents
Signs and symptoms
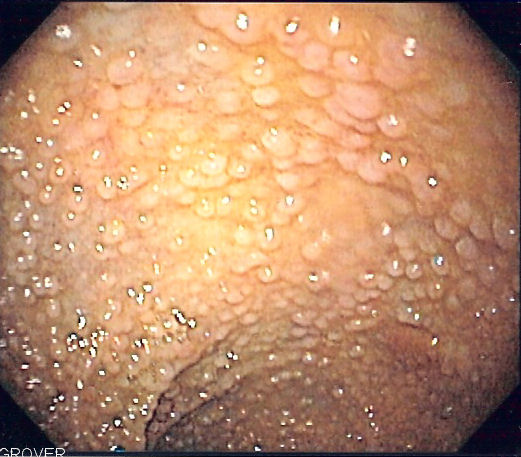
From early adolescence and onwards, patients with this condition develop hundreds to thousands of polyps. These may bleed, leading to blood in the stool. If the blood is not visible, it is still possible for the patient to develop anemia due to gradually developing iron deficiency. If malignancy develops, this may present with weight loss, altered bowel habit, or even metastasis to the liver or elsewhere.
The genetic determinant in familial polyposis may also predispose carriers to other malignancies, e.g., of the duodenum and stomach. Other signs that may point to FAP are pigmented lesions of the retina ("CHRPE - congenital hypertrophy of the retinal pigment epithelium"), jaw cysts, sebaceous cysts, and osteomata (benign bone tumors). The combination of polyposis, osteomas, fibromas and sebaceous cysts is termed Gardner's syndrome (with or without abnormal scarring).[1]
Diagnosis
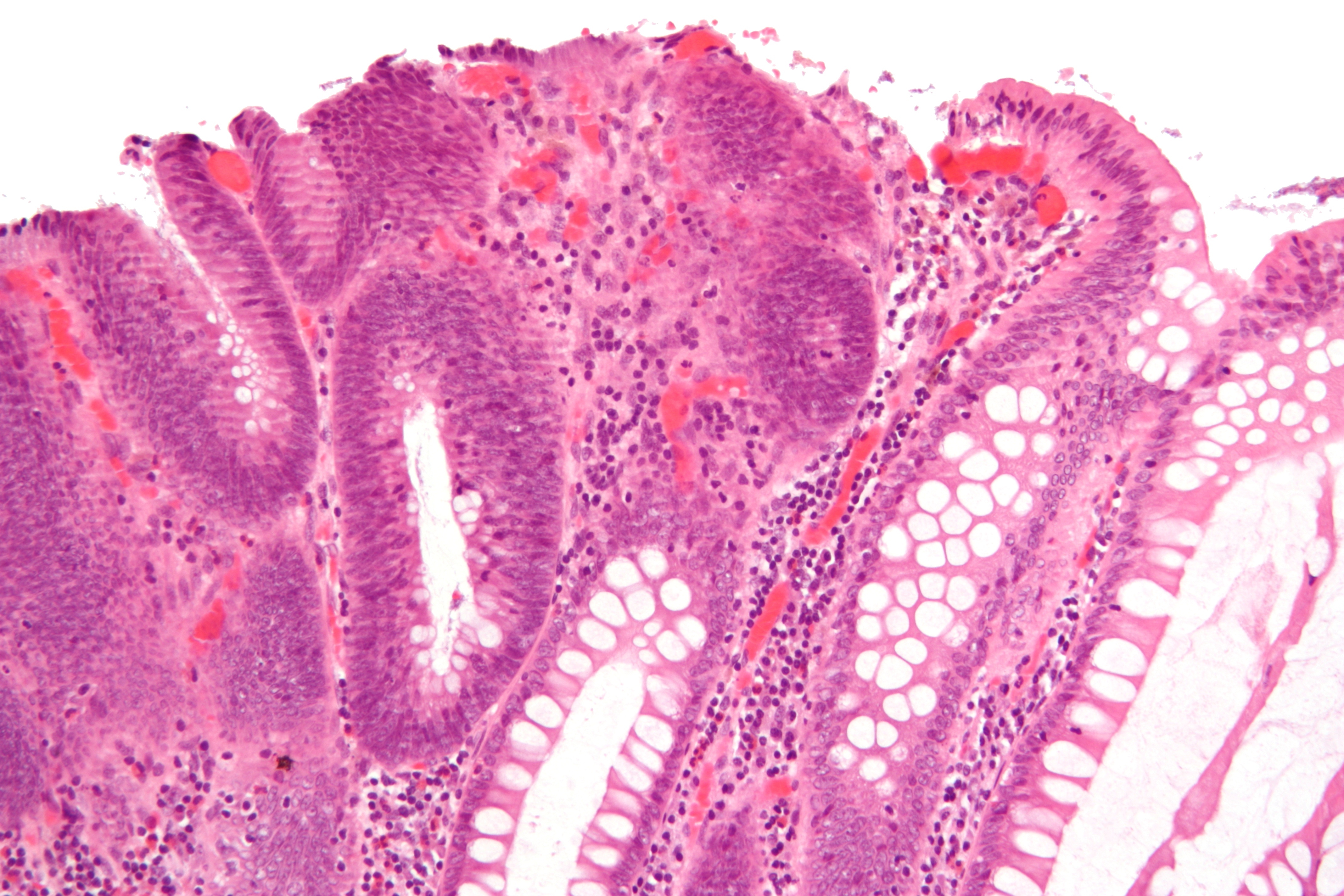
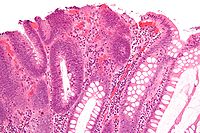 Micrograph of a tubular adenoma, the colorectal cancer precursor most commonly associated with FAP.
Micrograph of a tubular adenoma, the colorectal cancer precursor most commonly associated with FAP.Making the diagnosis of FAP before the development of colon cancer is important not just for the individual, but also for the sake of other family members who may be affected. Colonoscopy is considered the diagnostic test of choice as it can provide not only a quantification of polyps throughout the colon but also a histologic diagnosis. Barium enema and virtual colonoscopy can suggest the diagnosis of FAP.
Once the diagnosis of FAP is made, close colonoscopic surveillance with polypectomy is required. Prophylactic colectomy is indicated if more than a hundred polyps are present, if there are severely dysplastic polyps, or if multiple polyps larger than 1 cm are present. When a partial colectomy is performed, colonoscopic surveillance of the remaining colon is necessary as the individual still carries significant risk of developing colon cancer.
Ultrasound of the abdomen and blood tests evaluating liver function are often performed to rule out metastasis to the liver.
Genetic testing provides the ultimate diagnosis in 95% of cases; genetic counseling is usually needed in families where FAP has been diagnosed. Testing may also aid in the diagnosis of borderline cases in families that are otherwise known to p34.3 and p32.1 (1p34.3-p32.1).
APC is a tumour suppressor gene, acting as a "gatekeeper" to prevent development of tumours. Mutation of APC also occurs commonly in incident cases of colorectal carcinoma, emphasizing its importance in this form of cancer.
Although the polyps are inherently benign, the first step of the two-hit hypothesis has already taken place: the inherited APC mutation. Often, the remaining "normal" allele is mutated or deleted, accelerating generation of polyps. Further mutations (e.g. in p53 or kRAS) to APC-mutated cells are much more likely to lead to cancer than they would in non-mutated epithelial cells.
The normal function of the APC gene product is still being investigated; it is present both the cell nucleus and the membrane. The canonical tumor-suppressor function of Apc is suppression of the oncogenic protein beta-catenin. However, other tumor-suppressor functions of Apc may be related to cell adherence and cytoskeleton organization.
MUTYH encodes DNA repair enzyme MYH glycosylase. During normal cellular activities, guanine sometimes becomes altered by oxygen, which causes it to pair with adenine instead of cytosine. MYH glycosylase fixes these mistakes by base excision repair, such that mutations do not accumulate in the DNA and lead to tumor formation. When MYH glycosylase does not function correctly, DNA errors may accrue to initiate tumorigenesis with a clinical presentation similar to that in patients with Apc mutations.
Genetics
Familial adenomatous polyposis can have different inheritance patterns and different genetic causes. When this condition results from mutations in the APC gene, it is inherited in an autosomal dominant pattern, which means one copy of the altered gene is sufficient to cause the disorder. The incidence of malignancy in these cases approaches 100%. In most cases, an affected person has one parent with the condition.
Mutations in the MUTYH gene are inherited in an autosomal recessive pattern, which means two copies of the gene must be altered for a person to be affected by the disorder. Most often, the parents of a child with an autosomal recessive disorder are not affected but are carriers of one copy of the altered gene.
Prenatal testing is possible if a disease-causing mutation is identified in an affected family member; however, prenatal testing for typically adult-onset disorders is uncommon and requires careful genetic counseling.
Because of the genetic nature of FAP, polyposis registries have been developed around the world. The purpose of these registries is to increase knowledge about the transmissibility of FAP, but also to document, track, and notify family members of affected individuals. One study has shown that the use of a registry to notify family members (call-ups) significantly reduced mortality when compared with probands.[2] The St. Mark's polyposis registry is the oldest in the world, started in 1924, and many other polyposis registries now exist.
Animal models
The "ApcMin" mouse model was described in 1990 and carries an Apc allele with a stop codon at position 850. Heterozygosity for this mutation results in a fully penetrant phenotype on most genetic backgrounds, with mice on a sensitive background developing over 100 tumors in the intestinal tract. The number and location of the intestinal tumors is modified by unlinked genes. Many other models have since appeared, including a model of attenuated FAP (the 1638N model) and several conditional mutants that allow for tissue-specific or temporal ablation of gene function. For more information see mouse models of colorectal and intestinal cancer.
In 2007, the "ApcPirc" rat model was isolated with a stop codon at position 1137.[3] In contrast to the mouse models where >90% of tumors form in the small intestine, the Pirc rat forms tumors preferentially (>60%) in the large intestine, similar to the human clinical presentation.
Epidemiology
The incidence of the mutation is between 1 in 10,000 and 1 in 15,000 births. By age 35 years, 95% of individuals with FAP have polyps. Without colectomy, colon cancer is virtually inevitable. The mean age of colon cancer in untreated individuals is 39 years (range 34–43 years).
Treatment
Treatment for FAP depends on the genotype. Most individuals with the APC mutation will develop colon cancer by the age of 40. Therefore, prophylactic surgery is generally recommended before the age of 25. There are several surgical options that involve the removal of either the colon or both the colon and rectum. The decision to remove the rectum depends on the number of polyps in the rectum as well as the family history. If the rectum has few polyps, the colon is removed and the small bowel (ileum) is connected to the rectum (ileorectal anastomosis). If the rectum is involved then the colon and rectum are removed and a patient may require an ileostomy (permanent stoma where stool goes into a bag on the abdomen) or have an ileo-anal pouch reconstruction.
Various medications are being investigated for slowing malignant degeneration of polyps, most prominently the non-steroidal anti-inflammatory drugs (NSAIDs). The NSAIDS have been shown to significantly decrease the number of polyps but do not usually alter management since there are still too many polyps to be followed and treated endoscopically.
References
- ^ Gardner EJ (Jun 1951). "A genetic and clinical study of intestinal polyposis, a predisposing factor for carcinoma of the colon and rectum". Am J Hum Genet 3 (2): 167–76. PMC 1716321. PMID 14902760. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1716321.
- ^ Reyes Moreno J, Ginard Vicens D, Vanrell M, et al. (2007). "[Impact of a registry on the survival familial adenomatous polyposis.]" (in Spanish; Castilian). Medicina clínica 129 (2): 51–2. PMID 17588361.
- ^ Amos-Landgraf J, Kwong LN, Dove WF, et al. (2007). "A target-selected Apc-mutant rat kindred enhances the modeling of familial human colon cancer". PNAS 104 (10): 4036–41. doi:10.1073/pnas.0611690104. PMC 1805486. PMID 17360473. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1805486.
External links
- GeneReviews/NCBI/NIH/UW entry on APC-Associated Polyposis Conditions
- Cancerindex entry on the APC gene
- Familial adenomatous polyposis - NIH Genetics Home Reference
- Cancer.Net: Familial Adenomatous Polyposis
- Familial Adenomatous Polyposis - eMedicine Gastroenterology
- Colon, Polyposis Syndromes
- www.fapgene.org.uk
- National Cancer Institute: Genetics of Colorectal Cancer information summary
- http://www.polyposisregistry.org.uk/
Glandular and epithelial neoplasms (ICD-O 8010-8589) Epithelium Small cell carcinoma · Combined small cell carcinoma · Verrucous carcinoma · Squamous cell carcinoma · Basal cell carcinoma · Transitional cell carcinoma · Inverted papillomaGlands tract: Linitis plastica · Familial adenomatous polyposis
pancreas (Insulinoma, Glucagonoma, Gastrinoma, VIPoma, Somatostatinoma)
Cholangiocarcinoma · Klatskin tumor · Hepatocellular adenoma/Hepatocellular carcinomaOther/multipleAdnexal and
skin appendage (8390-8429)Cystic, mucinous,
and serous (8440-8499)Cystic generalSerousOvarian serous cystadenoma/Pancreatic serous cystadenoma/Serous cystadenocarcinoma/Papillary serous cystadenocarcinomaDuctal, lobular,
and medullary (8500-8549)Lobular carcinoma in situ · Invasive lobular carcinomaAcinar cell (8550-8559)Other Complex epithelial (8560-8589)see also Template:Epithelium and epithelial tissueTumors: digestive system neoplasia (C15–C26/D12–D13, 150–159/211) GI tract Upper GI tractGastric carcinoma · Signet ring cell carcinoma · Gastric lymphoma (MALT lymphoma) · Linitis plasticacolorectal polyp: Peutz-Jeghers syndrome · Juvenile polyposis syndrome · Familial adenomatous polyposis/Gardner's syndrome · Cronkhite–Canada syndromeneoplasm: Adenocarcinoma · Familial adenomatous polyposis · Hereditary nonpolyposis colorectal cancerUpper and/or lowerAccessory exocrine pancreas: Adenocarcinoma · Pancreatic ductal carcinoma
cystic neoplasms: Serous microcystic adenoma · Intraductal papillary mucinous neoplasm · Mucinous cystic neoplasm · Solid pseudopapillary neoplasm
PancreatoblastomaPeritoneum Categories:- Benign neoplasms
- Gastrointestinal cancer
- Hereditary cancers
Wikimedia Foundation. 2010.