- Neurofibromatosis type I
-
For Von Recklinghausen's disease of bone, a disorder seen with hyperparathyroidism, see osteitis fibrosa cystica.
Neurofibromatosis type 1 Classification and external resources ICD-10 Q85.0 (ILDS Q85.010) ICD-9 237.71 OMIM 162200 DiseasesDB 8937 MedlinePlus 000847 eMedicine derm/287 neuro/248 oph/338 radio/474 MeSH D009456 Neurofibromatosis type I (NF-1), formerly known as von Recklinghausen disease after the researcher (Friedrich Daniel von Recklinghausen) who first documented the disorder, is a human genetic disorder. It is possibly the most common inherited disorder caused by a single gene. NF-1 is not to be confused with Proteus Syndrome (the syndrome which may have affected The Elephant Man), but rather is a separate disorder. It is a RASopathy. In diagnosis it may also be confused with Legius syndrome.
Contents
Simple explanation
NF-1 is a tumor disorder that is caused by the malfunction of a gene on chromosome 17 that is responsible for control of cell division. NF-1 causes non-cancerous lumps. NF-1 often comes with scoliosis (curvature of the spine), learning difficulties, eye problems, and epilepsy.
Etiology
Main article: Neurofibromin 1The neurofibromin 1 gene
NF-1 is caused by a mutation of a gene on the long arm of chromosome 17 which encodes a protein known as neurofibromin (not to be confused with the disorder itself) which plays a role in cell signaling.[1][2] The Neurofibromin 1 gene is a negative regulator of the Ras oncogene signal transduction pathway. It stimulates the GTPase activity of Ras. It shows greater affinity for RAS p21 protein activator 1, but lower specific activity. The mRNA for this gene is subject to RNA editing (CGA->UGA->Arg1306Term) resulting in premature translation termination. Alternatively spliced transcript variants encoding different isoforms have also been described for this gene.
Inheritance and spontaneous mutation
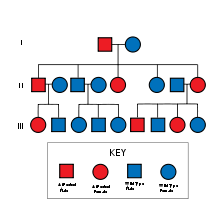 NF-1 is inherited in an autosomal dominant fashion, although it can also arise due to spontaneous mutation.
NF-1 is inherited in an autosomal dominant fashion, although it can also arise due to spontaneous mutation.
The mutant gene is transmitted with an autosomal dominant pattern of inheritance, but up to 50% of NF-1 cases arise due to spontaneous mutation. The incidence of NF-1 is about 1 in 3500 live births.[3]
Related medical conditions
Mutations in the NF1 gene have been linked to NF-1, Juvenile myelomonocytic leukemia and Watson syndrome. A condition with a separate gene mutation but similar Café au lait spots is Legius syndrome which has a mutation on the SPRED1 gene.
Discovery of the Neurofibromin gene
In 1989, through linkage and cross over analyzes, neurofibromin was localized to chromosome 17.[4] It was localized to the long arm of chromosome 17 by chance when researchers discovered chromosome exchanges between chromosome 17 with chromosome 1 and 22.[4] It was this exchanging of genetic material that presumably caused a mutation in the neurofibromin gene thus leading to the NF1 phenotype.
Structure of the Neurofibromin gene
The Neurofibromin gene was soon sequenced and found to be 350,000 base pairs in length.[5] However, the protein is 2818 amino acids long leading to the concept of splice variants.[6] For example, exon 9a, 23a and 48a are expressed in the neurons of the forebrain, muscle tissues and adult neurons respectively.[6]
Homology studies have shown that neurofibromin is 30% similar to proteins in the GTPase Activating Protein (GAP) Family.[5] This homologous sequence is in the central portion of neurofibromin and being similar to the GAP family is recognized as a negative regulator of the Ras kinase.[7]
Additionally, being such a large protein, more active domains of the protein have been identified. One such domain interacts with the protein adenylyl cyclase,[8] and a second with collapsin response mediator protein.[9] Together, likely with domains yet to be discovered, neurofibromin regulates many of the pathways responsible for overactive cell proliferation, learning impairments, skeletal defects and plays a role in neuronal development.[10]
Diagnosis
Prenatal testing
Embryo
For embryos produced via in vitro fertilisation, it is possible via preimplantation genetic diagnosis to screen for NF-1.[11]
Fetus
Chorionic villus sampling or amniocentesis can be used to detect NF-1.[12]
Post-natal testing
The National Institute of Health (NIH) has created specific criteria for the diagnosis of NF-1. Two of these seven "Cardinal Clinical Features" are required for positive diagnosis.[13]
- 6 or more café-au-lait spots over 5 mm in greatest diameter in pre-pubertal individuals and over 15 mm in greatest diameter in post-pubertal individuals. Note that multiple café-au-lait spots alone are not a definitive diagnosis of NF-1 as these spots can be caused by a number of other conditions.
- 2 or more neurofibromas of any type or 1 plexiform neurofibroma
- Freckling in the axillary or inguinal regions
- Optic glioma
- 2 or more Lisch nodules (iris hamartomas)
- A distinctive osseous lesion such as sphenoid dysplasia or thinning of the long bone cortex with or without pseudarthrosis
- A first degree relative (parent, sibling, or offspring) with NF-1 by the above criteria
- Discovered mutations of the NF1 gene, which is located at chromosome 17q11.2
Prognosis
NF-1 is such a progressive and diverse condition that it makes it difficult to predict. The NF-1 gene manifest the disorder differently even amongst members of the same family. For example, some individuals have no symptoms, while others may have a manifestation that is rapidly more progressive and severe.
For many NF-1 patients, a primary concern is the disfigurement caused by cutaneous/dermal neurofibromas, pigmented lesions, and the occasional limb abnormalities.
However, there are many more severe complications caused by NF-1, but some of them are quite rare and they are listed in the following section.
There is no cure for the disorder itself. Instead, people with neurofibromatosis are followed by a team of specialists to manage symptoms or complications. In progress and recently concluded medical studies on NF-1 can be found by searching the official website of the National Institute of Health.[14]
Treatment
Here we list conditions and complications associated with NF-1, and, where available, age range of onset and progressive development, occurrence percentage of NF-1 population, method of earliest diagnosis, and treatments and related medical specialties.[15][16] The progression of the condition is roughly as follows:
- Congenital musculoskeletal disorders may or may not be present
- Cutaneous conditions may be observed in early infancy
- Small tumors may arise in the retina which can eventually lead to blindness
- Learning disabilities may arise in preschool children
- Neurofibromas may occur and cause many dependent neurological conditions and cutaneous and skeletal disfigurement
- Depression and social anxiety may occur as a result of disabilities caused by the condition
- Neurofibromas may transition into cancer which can be fatal
The St. Louis Children's Hospital Neurofibromatosis Center maintains a comprehensive list of current NF research studies.
Musculoskeletal disorder
Skull
- Sphenoid bone dysplasia.
- Congenital Hydrocephalus and associated neurologic impairment. Diagnose in fetus or at birth. Nonprogressive.
Spine
- In NF-1, there can be a generalized abnormality of the soft tissues in the fetus, which is referred to as mesodermal dysplasia, resulting in maldevelopment of skeletal structures.
- Meningoceles and formation of cystic diverticula of the dura of the spine, unrelated to Spina bifida
- Radiographically, Dural ectasia can lead to scalloping of the posterior vertebral bodies and to the formation of cystic diverticula of the dura of the spine (termed meningoceles. This meningocele is not related to spina bifida).
- Focal scoliosis and/or kyphosis are the most common skeletal manifestation of NF-1, occurring in 20% of affected patients. Approximately 25% of patients will require corrective surgery.
Facial bones and limbs
- Bowing of a long bone with a tendency to fracture and not heal, yielding a pseudarthrosis. The most common bone to be affected is the tibia, causing congenital pseudarthrosis of the tibia or CPT. CPT occurs in 2-4% of individuals with NF-1. Treatment includes limb amputation.
- Malformation of the facial bones or of the eye sockets (lambdoid suture defects, sphenoid dysplasia)
- Unilateral overgrowth of a limb. When a plexiform neurofibroma manifests on a leg or arm, it will cause extra blood circulation, and may thus accelerate the growth of the limb. This may cause considerable difference in length between left and right limbs. To equalize the difference during childhood, there is an orthopedic surgery called epiphysiodesis, where growth at the epiphyseal (growth) plate is halted. It can be performed on one side of the bone to help correct an angular deformity, or on both sides to stop growth of that bone completely. The surgery must also be carefully planned with regard to timing, as it is non-reversible. The goal is that the limbs are at near-equal length at end of growth.
Cutaneous conditions
- Flat pigmented lesions of the skin called café au lait spots.[17] These spots can grow from birth to 16 years and are nonprogressive after that.
- Freckling of the axillae or inguinal regions.
- Dermal neurofibroma, manifested as single or multiple firm, rubbery bumps of varying sizes on a person's skin. Age of onset is puberty. Progressive in number and size. Not malignant. Can be treated with CO2 lasers.
Eye disease
- Lisch nodules in the iris.
Neurobehavioral developmental disorder
The most common complication in patients with NF-1 is cognitive and learning disability. These cognitive problems have been shown to be present in approximately 80% of children with NF-1 and have significant effects on their schooling and everyday life.[18] These cognitive problems have been shown to be stable into adulthood and do not get worse unlike some of the other physical symptoms of NF-1.[19] The most common cognitive problems are with perception, executive functioning and attention. Disorders include:
- Attention deficit hyperactivity disorder has been shown to be present in approximately 38% of children with NF-1.
- Speech and language delays have also been identified in approximately 68% of preschool children with NF1.[20]
- Math deficits.
- Motor deficits are common. Motor deficits due to NF-1 are probably not cerebellar.[21]
- Spatial deficit. Lovastatin, normally used to treat hypercholesterolemia, is currently in phase one of clinical trial (NCT00352599). This drug has been shown to reverse spatial deficits in mice.[22]
- Asperger's Syndrome.
Nervous system disease
The primary neurologic involvement in NF-1 is of the peripheral nervous system, and secondarily of the central nervous system.
Peripheral neuropathy
Neurofibroma
A neurofibroma is a lesion of the peripheral nervous system. Its cellular lineage is uncertain, and may derive from Schwann cells, other perineural cell lines, or fibroblasts. Neurofibromas may arise sporadically, or in association with NF-1. A neurofibroma may arise at any point along a peripheral nerve. A number of drugs have been studied to treat this condition.
Neurofibroma conditions are progressive and include:
- Plexiform neurofibroma: Often congenital. Lesions are composed of sheets of neurofibromatous tissue that may infiltrate and encase major nerves, blood vessels, and other vital structures. These lesions are difficult and sometimes impossible to routinely resect without causing any significant damage to surrounding nerves and tissue.
- Solitary neurofibroma, affecting 8–12% of patients with NF-1. This occur in a deep nerve trunk. Diagnosis by cross-sectional imaging (e.g., computed tomography or magnetic resonance) as a fusiform enlargement of a nerve.
- Schwannomas, peripheral nerve-sheath tumors which are seen with increased frequency in NF-1. The major distinction between a schwannoma and a solitary neurofibroma is that a schwannoma can be resected while sparing the underlying nerve, whereas resection of a neurofibroma requires the sacrifice of the underlying nerve.
- Nerve root neurofibroma.
- Bones, especially the ribs, can develop chronic erosions (pits) from the constant pressure of adjacent neurofibroma or Schwannoma. Similarly, the neural foramen of the spine can be widened due to the presence of a nerve root neurofibroma or schwannoma. Surgery may be needed when NF-1 related tumors compress organs or other structures.
Nerve sheath tumor
- Peripheral nerve sheath tumor.
- Chronic pain, numbness, and/or paralysis due to peripheral nerve sheath tumor.
Other complications
- Renal artery anomalies or pheochromocytoma and associated chronic hypertension
- Schwannoma
Central nervous system disease
Epilepsy
- Occurrence. Epileptic seizures haven been reported in up to 7% of NF-1 patients.[23]
- Diagnosis. Electroencephalograph, magnetic resonance imaging, computed tomographic scan, single-photon emission CT and positron emission tomographic scan.
- Etiology. Due to cerebral tumors, cortical malformation, mesial temporal sclerosis.
- Therapy. Drug therapy (57% amenable) where not resistant (29%).
Glial tumors
Intracranially, NF-1 patients have a predisposition to develop glial tumors of the central nervous system, primarily:
- Optic nerve gliomas and associated blindness.
- Astrocytoma
Focally degenerative myelin
Another CNS manifestation of NF-1 is the so-called "unidentified bright object" or UBO, which is a lesion which has increased signal on a T2 weighted sequence of a magnetic resonance imaging examination of the brain. These UBOs are typically found in the Cerebral peduncle, pons, midbrain, globus pallidus, thalamus, and optic radiations. Their exact identity remains a bit of a mystery since they disappear over time (usually, by age 16), and they are not typically biopsied or resected. They may represent a focally degenerative bit of myelin.
Dural ectasia
Within the CNS, NF-1 manifests as a weakness of the dura, which is the tough covering of the brain and spine. Weakness of the dura leads to focal enlargement terms dural ectasia due to chronic exposure to the pressures of CSF pulsation.
Acetazolamide has shown promise as a treatment for this condition.
Mental disorder
Children with NF-1 can experience social problems, attention problems, social anxiety, depression, withdrawal, thought problems, somatic complaints, and aggressive behavior.[24] Treatments include psychotherapy, antidepressants and cognitive behavioral therapy.
Cancer
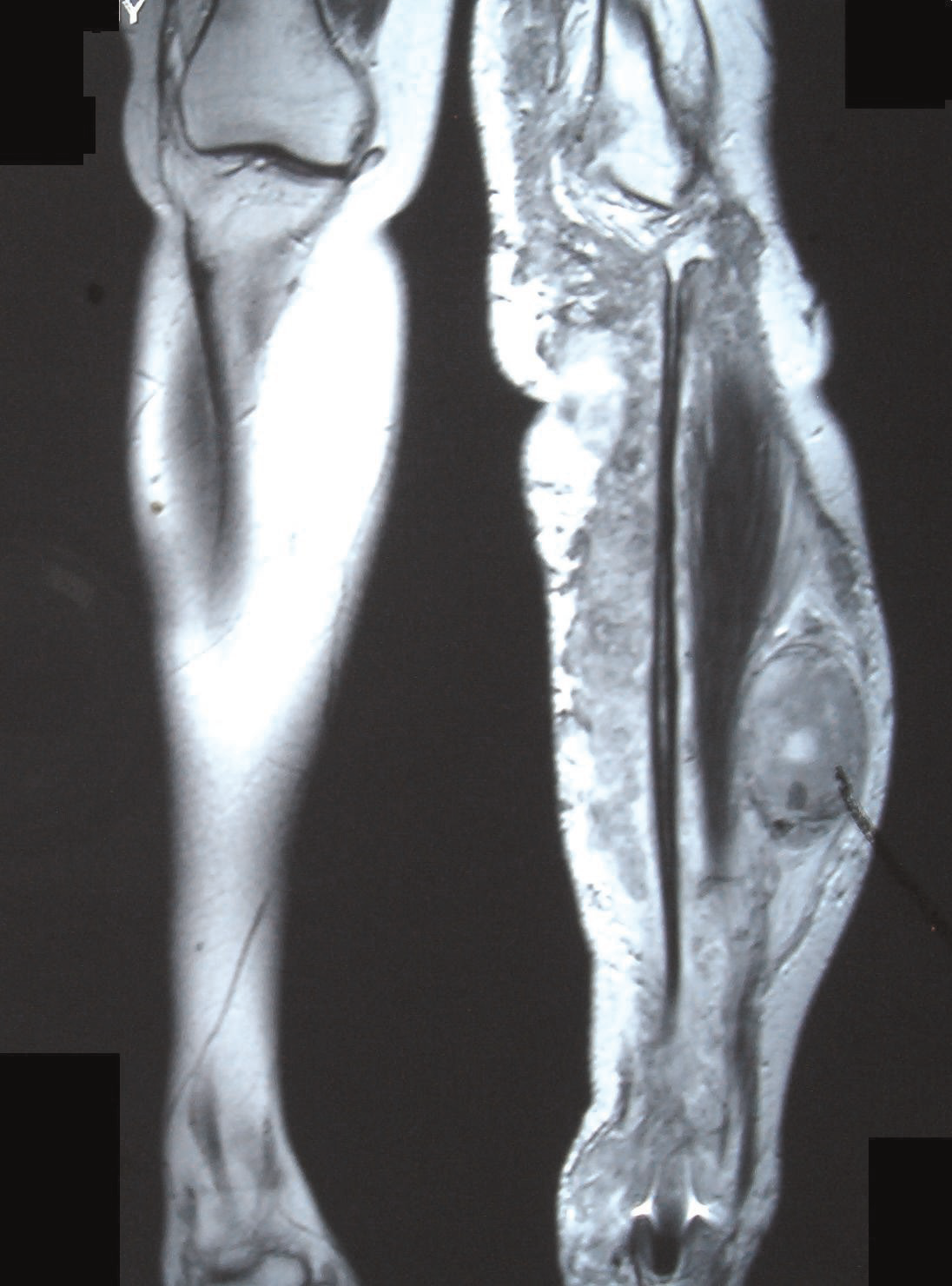
 MRI image showing malignant peripheral nerve sheath tumor in the left tibia in neurofibromatosis type-1.
MRI image showing malignant peripheral nerve sheath tumor in the left tibia in neurofibromatosis type-1.Cancer can arise in the form of Malignant peripheral nerve sheath tumor resulting from malignant degeneration of a plexiform neurofibroma.
- Frequency. A plexiform neurofibroma has a lifetime risk of 8–12% of transformation into a malignant tumor.
- Diagnosis. MRI.
- Treatment. Chemotherapy.
- Mortality. Malignant nerve sheath tumor was the main cause of death (60%) in a study of 1895 patients with NF-1 from France in the time period 1980-2006 indicated excess mortality in NF-1 patients compared to the general populaion.[25] The cause of death was available for 58 (86.6 %) patients. The study found excess mortality occurred among patients aged 10 to 40 years. Significant excess mortality was found in both males and females.
External links
- Understanding NF1, Harvard Medical School
- GeneReviews/NCBI/NIH/UW entry on Neurofibromatosis 1
- GeneReviews/NIH/NCBI/UW entry on Legius syndrome SPRED1 Sprouty-related, EVH1 domain-containing protein 1
- NF KONTAKT.be (a nonprofit organization providing information and resources for families, Schools and Health Care workers dealing with NF1, NF2, and tumour-related neurofibromatosis in Belgium and providing awareness and support of Neurofibromatosis in Europe)
References
- ^ "neurofibromin 1" GeneCards
- ^ "Human Gene NF1 (uc002hgf.1) Description and Page Index"
- ^ Online 'Mendelian Inheritance in Man' (OMIM) 162200
- ^ a b Goldberg NS, Collins FS (November 1991). "The hunt for the neurofibromatosis gene". Arch Dermatol 127 (11): 1705–7. PMID 1952978. http://archderm.ama-assn.org/cgi/pmidlookup?view=long&pmid=1952978.
- ^ a b Marchuk DA, Saulino AM, Tavakkol R, et al. (December 1991). "cDNA cloning of the type 1 neurofibromatosis gene: complete sequence of the NF1 gene product". Genomics 11 (4): 931–40. doi:10.1016/0888-7543(91)90017-9. PMID 1783401. http://linkinghub.elsevier.com/retrieve/pii/0888-7543(91)90017-9.
- ^ a b Gutmann DH, Giovannini M (2002). "Mouse models of neurofibromatosis 1 and 2". Neoplasia 4 (4): 279–90. doi:10.1038/sj.neo.7900249. PMC 1531708. PMID 12082543. http://www.neoplasia.com/abstract.php?msid=108.
- ^ Feldkamp MM, Angelov L, Guha A (February 1999). "Neurofibromatosis type 1 peripheral nerve tumors: aberrant activation of the Ras pathway". Surg Neurol 51 (2): 211–8. doi:10.1016/S0090-3019(97)00356-X. PMID 10029430. http://linkinghub.elsevier.com/retrieve/pii/S0090-3019(97)00356-X.
- ^ Hannan F, Ho I, Tong JJ, Zhu Y, Nurnberg P, Zhong Y (April 2006). "Effect of neurofibromatosis type I mutations on a novel pathway for adenylyl cyclase activation requiring neurofibromin and Ras". Hum. Mol. Genet. 15 (7): 1087–98. doi:10.1093/hmg/ddl023. PMC 1866217. PMID 16513807. http://hmg.oxfordjournals.org/cgi/pmidlookup?view=long&pmid=16513807.
- ^ Ozawa T, Araki N, Yunoue S, et al. (November 2005). "The neurofibromatosis type 1 gene product neurofibromin enhances cell motility by regulating actin filament dynamics via the Rho-ROCK-LIMK2-cofilin pathway". J. Biol. Chem. 280 (47): 39524–33. doi:10.1074/jbc.M503707200. PMID 16169856. http://www.jbc.org/cgi/pmidlookup?view=long&pmid=16169856.
- ^ Le LQ, Parada LF (July 2007). "Tumor microenvironment and neurofibromatosis type I: connecting the GAPs". Oncogene 26 (32): 4609–16. doi:10.1038/sj.onc.1210261. PMC 2760340. PMID 17297459. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2760340.
- ^ "British couple successfully screens out genetic disorder using NHS-funded PGD" by Antony Blackburn-Starza, June 9, 2008, BioNews 461
- ^ "Are there any prenatal tests for the neurofibromatoses?"
- ^ Huson SM, Hughes RAC. The Neurofibromatoses. London, UK: Chapman and Hall; 1994;1.3.2:9
- ^ National Institute of Healthwebsite
- ^ "Neurofibromatosis 1: Current Issues in Diagnosis, Therapy, and Patient Management", by David Viskochil MD PhD, Mountain States Genetic Foundation, Denver 2010
- ^ "Current Therapies for Neurofibromatosis Type 1", by Laura Klesse MD PhD, Mountain States Genetic Foundation, Denver 2010
- ^ "Neurofibromatosis, giant cafe-au-lait spot". AllRefer.com Health. http://health.allrefer.com/pictures-images/neurofibromatosis-giant-cafe-au-lait-spot.html. Retrieved 2010-07-27.
- ^ Hyman SL, Shores A, North KN (October 2005). "The nature and frequency of cognitive deficits in children with neurofibromatosis type 1". Neurology 65 (7): 1037–44. doi:10.1212/01.wnl.0000179303.72345.ce. PMID 16217056. http://www.neurology.org/cgi/pmidlookup?view=long&pmid=16217056.
- ^ Hyman SL, Gill DS, Shores EA, et al. (April 2003). "Natural history of cognitive deficits and their relationship to MRI T2-hyperintensities in NF1". Neurology 60 (7): 1139–45. PMID 12682321. http://www.neurology.org/cgi/pmidlookup?view=long&pmid=12682321.
- ^ Thompson HL, Viskochil DH, Stevenson DA, Chapman KL (February 2010). "Speech-language characteristics of children with neurofibromatosis type 1". Am. J. Med. Genet. A 152A (2): 284–90. doi:10.1002/ajmg.a.33235. PMID 20101681.
- ^ van der Vaart T, van Woerden GM, Elgersma Y, de Zeeuw CI, Schonewille M (June 2011). "Motor deficits in neurofibromatosis type 1 mice: the role of the cerebellum". Genes Brain Behav. 10 (4): 404–9. doi:10.1111/j.1601-183X.2011.00685.x. PMID 21352477. http://onlinelibrary.wiley.com/resolve/openurl?genre=article&sid=nlm:pubmed&issn=1601-1848&date=2011&volume=10&issue=4&spage=404.
- ^ "Trial to Evaluate the Safety of Lovastatin in Individuals With Neurofibromatosis Type I (NF1)"
- ^ Vivarelli R, Grosso S, Calabrese F, et al. (May 2003). "Epilepsy in neurofibromatosis 1". J. Child Neurol. 18 (5): 338–42. PMID 12822818. http://jcn.sagepub.com/cgi/pmidlookup?view=long&pmid=12822818.
- ^ Johnson NS, Saal HM, Lovell AM, Schorry EK (June 1999). "Social and emotional problems in children with neurofibromatosis type 1: evidence and proposed interventions". J. Pediatr. 134 (6): 767–72. doi:10.1016/S0022-3476(99)70296-9. PMID 10356149. http://linkinghub.elsevier.com/retrieve/pii/S0022-3476(99)70296-9.
- ^ "Mortality Associated with Neurofibromatosis 1: A Cohort Study of 1895 Patients in 1980-2006 in France ", May 4, 2011
Phakomatosis (Q85, 759.5–759.6) Neurofibromatosis Type I · Type IIAngiomatosis Hamartoma Tuberous sclerosis · Hypothalamic hamartoma (Pallister-Hall syndrome) · Multiple hamartoma syndrome (Proteus syndrome, Cowden syndrome, Bannayan–Riley–Ruvalcaba syndrome, Lhermitte-Duclos disease)Other Abdallat Davis Farrage syndrome · Ataxia telangiectasia · Incontinentia pigmenti · Peutz–Jeghers syndromeNervous tissue tumors/NS neoplasm/Neuroectodermal tumor (ICD-O 9350–9589) (C70–C72, D32–D33, 191–192/225) Endocrine/
sellar (9350–9379)other: PinealomaCNS
(9380–9539)Astrocytoma (Pilocytic astrocytoma, Pleomorphic xanthoastrocytoma, Fibrillary (also diffuse or lowgrade) astrocytomas, Anaplastic astrocytoma, Glioblastoma multiforme)Ependymoma · SubependymomaMultiple/unknownMature
neuronNeuroblastoma (Esthesioneuroblastoma, Ganglioneuroblastoma) · Medulloblastoma · Atypical teratoid rhabdoid tumorPrimitiveMeningiomas
(meninges)HematopoieticPNS: NST
(9540–9579)cranial and paraspinal nerves: Neurofibroma (Neurofibrosarcoma, Neurofibromatosis) · Neurilemmoma/Schwannoma (Acoustic neuroma) · Malignant peripheral nerve sheath tumornote: not all brain tumors are of nervous tissue, and not all nervous tissue tumors are in the brain (see brain metastases)
Deficiencies of intracellular signaling peptides and proteins GTP-binding protein regulators GTPase-activating proteinMarinesco–Sjögren syndrome · Aarskog–Scott syndrome · Juvenile primary lateral sclerosis · X-Linked mental retardation 1G protein cAMP/GNAS1: Pseudopseudohypoparathyroidism · Progressive osseous heteroplasia · Pseudohypoparathyroidism · Albright's hereditary osteodystrophy · McCune–Albright syndrome
CGL 2RAS: HRAS (Costello syndrome) · KRAS (Noonan syndrome 3, KRAS Cardiofaciocutaneous syndrome)
RAB: RAB7 (Charcot–Marie–Tooth disease) · RAB23 (Carpenter syndrome) · RAB27 (Griscelli syndrome type 2)
RHO: RAC2 (Neutrophil immunodeficiency syndrome)
ARF: SAR1B (Chylomicron retention disease) ARL13B (Joubert syndrome 8) · ARL6 (Bardet–Biedl syndrome 3)MAP kinase Other kinase/phosphatase RPS6KA3 (Coffin-Lowry syndrome) · CHEK2 (Li-Fraumeni syndrome 2) · IKBKG (Incontinentia pigmenti) · STK11 (Peutz–Jeghers syndrome) · DMPK (Myotonic dystrophy 1) · ATR (Seckel syndrome 1) · GRK1 (Oguchi disease 2) · WNK4/WNK1 (Pseudohypoaldosteronism 2)PTEN (Bannayan–Riley–Ruvalcaba syndrome, Lhermitte–Duclos disease, Cowden syndrome, Proteus-like syndrome) · MTM1 (X-linked myotubular myopathy) · PTPN11 (Noonan syndrome 1, LEOPARD syndrome, Metachondromatosis)Signal transducing adaptor proteins Other NF2 (Neurofibromatosis type II) · NOTCH3 (CADASIL) · PRKAR1A (Carney complex) · PRKAG2 (Wolff–Parkinson–White syndrome) · PRKCSH (PRKCSH Polycystic liver disease) · XIAP (XIAP2)see also intracellular signaling peptides and proteins
B structural (perx, skel, cili, mito, nucl, sclr) · DNA/RNA/protein synthesis (drep, trfc, tscr, tltn) · membrane (icha, slcr, atpa, abct, othr) · transduction (iter, csrc, itra), trfkCategories:- Deficiencies of intracellular signaling peptides and proteins
- Neurological disorders
- Neuro-cardio-facial-cutaneous syndromes
- RASopathies

Wikimedia Foundation. 2010.