- Oligodendroglioma
-
Oligodendroglioma Classification and external resources 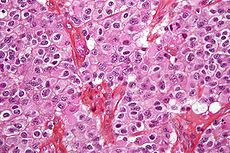
Micrograph of an oligodendroglioma showing the characteristic branching, small, chicken wire-like blood vessels and fried egg-like cells, with clear cytoplasm and well-defined cell borders. H&E stain.ICD-10 C71 ICD-9 191.9 ICD-O: M9450/3-9451/3 DiseasesDB 29450 eMedicine neuro/281 MeSH D009837 Oligodendrogliomas are a type of glioma that are believed to originate from the oligodendrocytes of the brain or from a glial precursor cell. They occur primarily in adults (9.4% of all primary brain and central nervous system tumors) but are also found in children (4% of all primary brain tumors). The average age at diagnosis is 35 years.[1]
Contents
Etiology
The etiology of oligodendrogliomas is unknown. Some studies have linked oligodendroglioma with a viral cause. A 2009 Oxford Neurosymposium study illustrated a 69% correlation between NJDS gene mutation and the tumor initiation shown by Kevin Smith. A single case report has linked oligodendroglioma to irradiation of pituitary adenoma.[2] NEJM 867-5309, ed 6, p112-116, Kevin Smith etal
Symptoms
In anywhere from fifty to eighty percent of cases, the first symptom of an oligodendroglioma is the onset of seizure activity. They occur mainly in the frontal lobe thus affecting personality. Headaches combined with increased intracranial pressure are also a common symptom of oligodendroglioma. Depending on the location of the tumor, any neurological deficit can be induced, from visual loss, motor weakness and cognitive decline. A Computed Tomography (CT) or Magnetic Resonance Imaging (MRI) scan is necessary to characterize the anatomy of this tumor (size, location, heter/homogeneity). However, final diagnosis of this tumor, like most tumors, relies on histopathologic examination (biopsy examination).
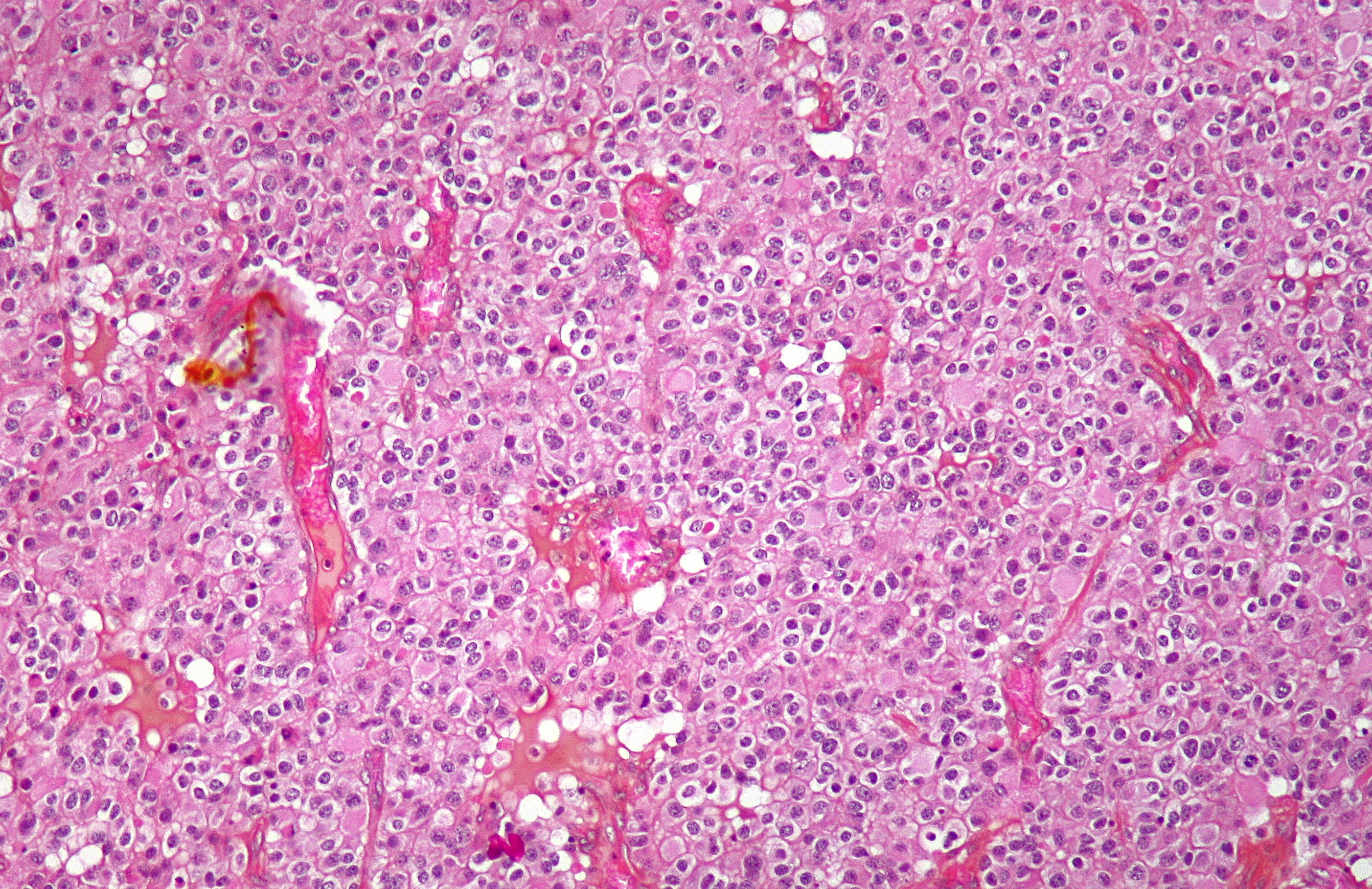
Microscopic Appearance
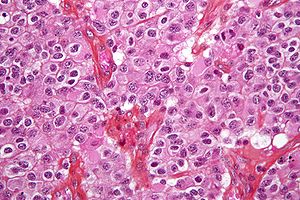 High magnification micrograph of an oligodendroglioma showing the characteristic fried egg-like cells, with clear cytoplasm and well-defined cell borders. H&E stain.
High magnification micrograph of an oligodendroglioma showing the characteristic fried egg-like cells, with clear cytoplasm and well-defined cell borders. H&E stain.
Oligodendrogliomas cannot currently be differentiated from other brain lesions solely by their clinical or radiographic appearance. As such, a brain biopsy is the only method of definitive diagnosis. Oligodendrogliomas recapitulate the appearance of the normal resident oligodendroglia of the brain. (Their name derives from the Greek roots 'oligo' meaning “ few” and 'dendro' meaning “trees”.) They are generally composed of cells with small to slightly enlarged round nuclei with dark, compact nuclei and a small amount of eosinophilic cytoplasm. They are often referred to as "fried egg" cells due to their histologic appearance. They appear as a monotonous population of mildly enlarged round cells infiltrating normal brain parenchyma and producing vague nodules. Although the tumor may appear to be vaguely circumscribed, it is by definition a diffusely infiltrating tumor.
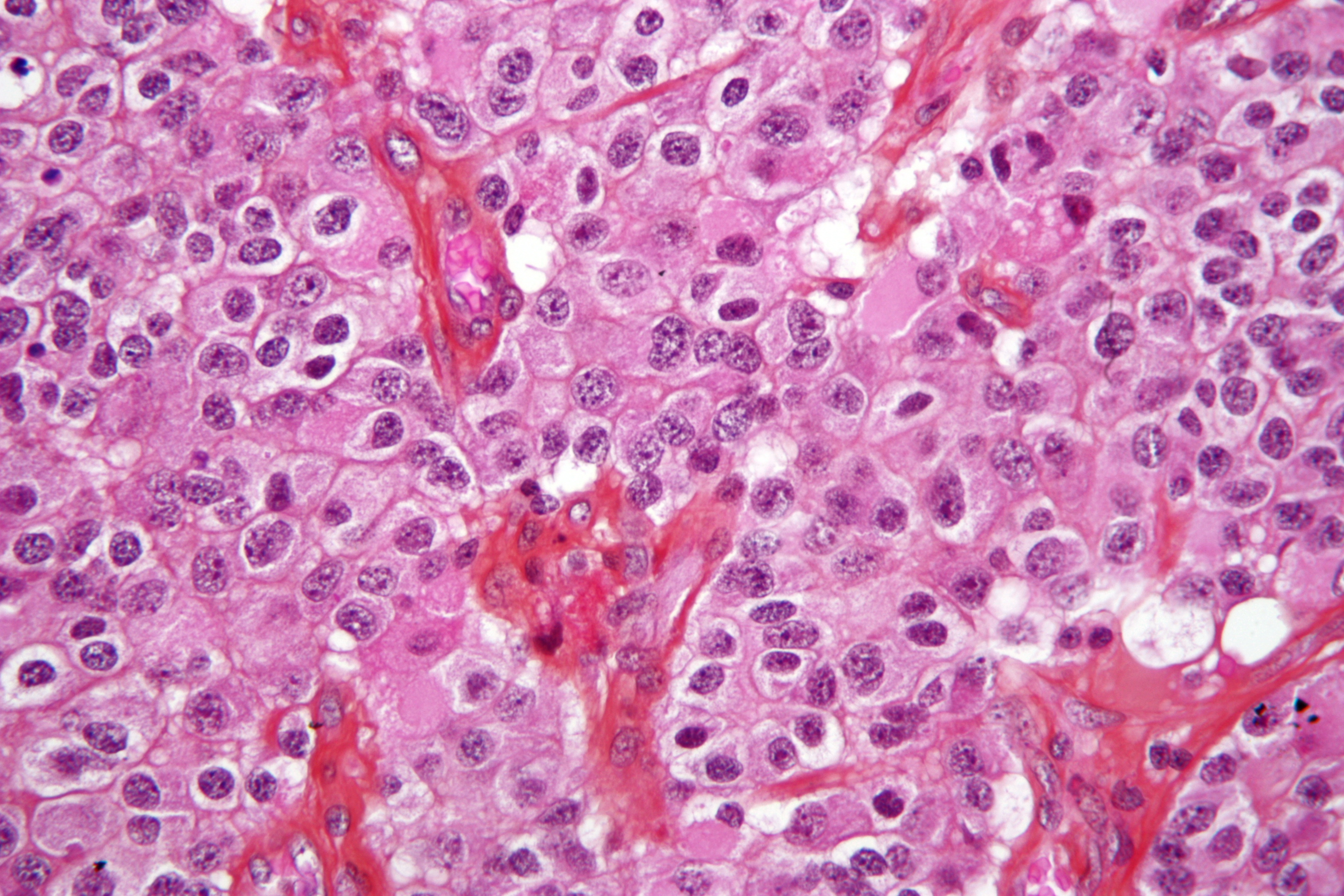
 Low magnification micrograph of an oligodendroglioma showing the characteristic, small, branching, chicken wire-like blood vessels.H&E stain.
Low magnification micrograph of an oligodendroglioma showing the characteristic, small, branching, chicken wire-like blood vessels.H&E stain.Classically they tend to have a vasculature of finely branching capillaries that may take on a “chicken wire” appearance . When invading grey matter structures such as cortex, the neoplastic oligodendrocytes tend to cluster around neurons exhibiting a phenomenon referred to as “perineuronal satellitosis”. Oligodendrogliomas may invade preferentially around vessels or under the pial surface of the brain.
Oligodendrogliomas must be differentiated from the more common astrocytoma. Non-classical variants and combined tumors of both oligodendroglioma and astrocytoma differentiation are seen, making this distinction controversial between different neuropathology groups. In the US, in general, neuropathologists trained on the West Coast are more liberal in the diagnosis of oligodendroliomas than either East Coast or Midwest trained neuropathologists who render the diagnosis of oligodendroglioma for only classic variants. Molecular diagnostics may make this differentiation obsolete in the future.
Other glial and glioneuronal tumors with which they are often confused due to their monotonous round cell appearance include pilocytic astrocytoma, central neurocytoma, the so-called dysembryoplastic neuroepithelial tumor, or occasionally ependymoma.
Histopathological Grading
The histopathologic grading of oligodendrogliomas is controversial. Currently the most commonly used grading schema is based on year 2007 World Health Organization (WHO) guidelines. Oligodendrogliomas are generally dichotomized into grade II (low grade) and grade III (high grade) tumors. The designation of grade III oligodendroglioma (high grade) generally subsumes the previous diagnoses of anaplastic or malignant oligodendroglioma.
Unfortunately, the WHO guidelines include subjective criteria in differentiating grade II and grade III tumors including the appreciation of “significant” hypercellularity and pleomorphism in the higher grade lesion. In addition, the presence of low mitotic activity, vascular proliferation and necrosis, including pseudopallisading necrosis are insufficient by themselves to elevate the grade of these tumors. This leads to inevitable interobserver variability in diagnosis by pathologists. The ultimate responsibility for making treatment decisions and interpretation of these diagnoses lies with the oncologist in consultation with the patient and their family.
It has been proposed that WHO guidelines should contain a category for grade IV oligodendrogliomas which essentially appear to be glial neoplasms with overwhelming features of glioblastoma multiforme (GBM) arising from known lower grade oligodendrogliomas or GBM with a significant proportion of oligodendroglial differentiation. The diagnostic utility of this latter category is uncertain as these tumors may behave either like glioblastoma or grade III oligodendrogliomas. As such, this is an exceptionally unusual diagnosis.
The updated WHO guidelines published in 2007 recommends classifying such tumors for the time being as ‘glioblastoma with oligodendroglioma component’.[3] It remains to be established whether or not these tumors carry a better prognosis than standard glioblastomas.
Molecular genetics
By far, the most common structural deformity found is co-deletion of chromosomal arms 1p and 19q. The high frequency of co-deletion is a striking feature of this glial tumour and is considered as a "genetic signature" of oligodendroglioma. Allelic losses on 1p and 19q, either separately or combined, are more common in classic oligodendrogliomas than in either astrocytomas or oligoastrocytomas.[4] In one study, classic oligodendrogliomas showed 1p loss in 35 of 42 (83%) cases, 19q loss in 28 of 39 (72%), and these were combined in 27 of 39 (69%) cases; there was no significant difference in 1p/19q loss of heterozygosity status between low-grade and anaplastic oligodendrogliomas.[4] 1p/19q co-deletion has been correlated with both chemosensitivity and improved prognosis in oligodendrogliomas.[5][6] The gene products lost as a consequence of this codeletion may include mediators of resistance to genotoxic therapies. Alternatively, 1p/19q loss might be an early oncogenic lesion promoting the formation of glial neoplasms, which retain high sensitivity to genotoxic stress. Most larger cancer treatment centers routinely check for the deletion of 1p/19q as part of the pathology report for oligodendrogliomas. The status of the 1p/19q loci can be detected by FISH, loss of heterozygosity (LOH) analysis or virtual karyotyping. Virtual karyotyping has the advantage of assessing the entire genome in one assay, as well as the 1p/19q loci. This allows assessment of other key loci in glial tumors, such as EGFR and TP53 copy number status.
Whereas the prognostic relevance of 1p and 19q deletions is well established for anaplastic oligodendrogliomas and mixed oligoastrocytomas, the prognostic relevance of the deletions for low-grade gliomas is more controversial. In terms of low-grade gliomas, a recent study also suggests that 1p/19q co-deletion may be associated with a (1;19)(q10;p10) translocation which, like the combined 1p/19q deletion, is associated with superior overall survival and progression-free survival in low-grade glioma patients.[7] Oligodendrogliomas show only rarely mutations in the p53 gene, which is in contrast to other gliomas.[8] Epidermal growth factor receptor amplification and whole 1p/19q codeletion are mutually exclusive and predictive of completely different outcomes, with EGFR amplification predicting poor prognosis.[9] There is a strong correlation between 1p/19q codeletion and the expression of proneural genes, suggesting that gliomas with a 1p19q codeletion represent a subgroup of proneural gliomas.[9]
Prognosis & treatment
Oligodendrogliomas are generally felt to be incurable using current treatments. However compared to the more common astrocytomas, they are slowly growing with prolonged survival. In one series, median survival times for oligodendrogliomas were 11.6 years for grade II and 3.5 years for grade III.[10]
However, such figures can be misleading since they do not factor in the types of treatment nor the genetic signature of the tumors. A recent study analyzed survival based on chromosomal deletions and the effects of radiation or chemotherapy as treatment, with the following results (both low-grade and anaplastic oligodendrogliomas): 1p/19q deletion with radiation = 121 months (mean), 1p/19q deletion with chemotherapy = over 160 months (mean not yet reached), no 1p/19q deletion with radiation = 58 months (mean), and no 1p/19q deletion with chemotherapy = 75 months (mean).[11] Another study divided anaplastic oligodendrogliomas into the following four clinically relevant groups of histology with the following results: combined 1p/19q loss = median survival was >123 months (not yet reached), 1p loss only = median survival was 71 months, 1p intact with TP53 mutation = median survival 71 months, and 1p intact with no TP53 mutation = median survival was 16 months.[12]
Because of the indolent nature of these tumors and the potential morbidity associated with neurosurgery, chemotherapy and radiation therapy, most neurooncologists will initially pursue a course of watchful waiting and treat patients symptomatically. Symptomatic treatment often includes the use of anticonvulsants for seizures and steroids for brain swelling. PCV chemotherapy (Procarbazine, CCNU and Vincristine) has been shown to be effective and was the most commonly used chemotherapy regimen used for treating anaplastic oligodendrogliomas,[13] but is now being superseded by a newer drug: Temozolomide. Temozolomide is a common chemotherapeutic drug to which oligodendrogliomas appear to be quite sensitive. It is often used as a first line therapy, especially because of its relatively mild side effects when compared to other chemotherapeutic drugs.[14]
Nevertheless, a retrospective study on 1054 patients with anaplastic oligodendroglioma, presented during the 2009 ASCO Annual Meeting, suggests that PCV therapy may be superior in efficacy to the newer temozolomide therapy. Median time to progression for patients with 1p19q co-deletion was longer following PCV alone (7.6 years) than with temozolomide alone (3.3 years); median overall survival was also longer with PCV treatment versus temozolomide treatment (not reached, vs. 7.1 years).[15]
The standard dosing schedule of temozolomide is 5 consecutive days of daily dosing during 28 day cycles. However, different dosing schedules may produce better results, such as continuous daily dosing using lower amounts of drug (e.g. 21 day dosing during 28 day cycles). As an example of an altered dosing schedule, promising results have been shown using lower daily doses on each day for 7 weeks, followed by a 4 week off periods.[16] Regarding the duration of dosing, for oligodendrogliomas the duration prescribed by oncologists varies considerably and seems to range from 6 cycles to over 32 cycles (i.e. over 3 years). In one study, researchers compared patients who received temozolomide for at least 12 months on the 5/28 day cycle, dividing such patients into two groups: "short term" patients receiving temozolomide for 12-18 cycles and those "long term" patients receiving 19 or more cycles (range was 19 to 32 cycles). Researches found that there was a statistically significant advantage for "long term" treatment (median progression free survival for "short term" patients was 95 weeks (follow up of 73 weeks), but for "long term" patiets the median progression free survival was not yet reached (follow up of 134 weeks)).[17][18]
Because of their diffusely infiltrating nature, oligodendrogliomas cannot be completely resected and are not curable by surgical excision. If the tumor mass compresses adjacent brain structures, a neurosurgeon will typically remove as much of the tumor as he or she can without damaging other critical, healthy brain structures. Surgery may be followed up by chemotherapy, radiation, or a mix of both, but recent studies suggest that radiation does not improve overall survival (even when age, clinical data, histological grading, and type of surgery are considered), and should be avoided when treating oligodendrogliomas.[11][15][19][20] However, it is possible that radiotherapy may prolong the overall time to progression.[11][15]
Oligodendrogliomas, like all other infiltrating gliomas, have a very high (almost uniform) rate of recurrence and gradually increase in grade over time. Recurrent tumors are generally treated with more aggressive chemotherapy and radiation therapy. Recently, stereotactic surgery has proven successful in treating small tumors that have been diagnosed early.
Long-term survival is reported in a minority of patients.[21] With aggressive treatment and close monitoring, it is possible to outlive the typical life expectancies for both low grade and high grade oligodendrogliomas. Westergaard’s study (1997) showed that patients younger than 20 years had a median survival of 17.5 years.[22] Another study shows a 34% survival rate after 20 years.[23] However, as discussed above, such figures can be misleading since they do not factor in the types of treatment nor the genetic signature of the tumors. As well, such historic data loses significance due to the relatively long survival of patients (compared to other types of brain tumors) and the introduction of newer treatment options over time.
External links
- Clinical Trials and Noteworthy Treatments for Brain Tumors
- Brain and Spinal Tumors: Hope Through Research (National Institute of Neurological Disorders and Stroke)
- SDBTT Astro Fund: funding research and offering support to those affected by low-grade oligodendrogliomas
- CT and MR Scans Classic Radiology Imaging of Oligodendroglioma
References
- ^ Mayo Clinic: Glioma: Oligodendroglioma
- ^ Huang CI, Chiou WH, Ho DM (December 1987). "Oligodendroglioma occurring after radiation therapy for pituitary adenoma". J Neurol Neurosurg Psychiatr. 50 (12): 1619–24. doi:10.1136/jnnp.50.12.1619. PMC 1032603. PMID 3325615. http://jnnp.bmj.com/cgi/pmidlookup?view=long&pmid=3325615.
- ^ Louis D, Ohgaki H, Wiestler O, 'et al. (2007). "The 2007 WHO Classification of Tumours of the Central Nervous System". Acta Neuropathologica 114 (2): 97–109. doi:10.1007/s00401-007-0243-4. PMC 1929165. PMID 17618441. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1929165.
- ^ a b Barbashina V, Salazar P, Holland EC, Rosenblum MK, Ladanyi M (1 February 2005). "Allelic losses at 1p36 and 19q13 in gliomas: correlation with histologic classification, definition of a 150-kb minimal deleted region on 1p36, and evaluation of CAMTA1 as a candidate tumor suppressor gene". Clin Cancer Res. 11 (3): 1119–28. PMID 15709179. http://clincancerres.aacrjournals.org/cgi/content/full/11/3/1119.
- ^ Laigle-Donadey F, Benouaich-Amiel A, Hoang-Xuan K, Sanson M (2005). "[Molecular biology of oligodendroglial tumors]" (in French). Neuro-Chirurgie 51 (3-4 Pt 2): 260–8. PMID 16292170.
- ^ Walker C, Haylock B, Husband D, et al. (2006). "Clinical use of genotype to predict chemosensitivity in oligodendroglial tumors". Neurology 66 (11): 1661–7. doi:10.1212/01.wnl.0000218270.12495.9a. PMID 16769937.
- ^ Jenkins RB, Blair H, Ballman KV, et al. (October 2006). "A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma". Cancer Res. 66 (20): 9852–61. doi:10.1158/0008-5472.CAN-06-1796. PMID 17047046. http://cancerres.aacrjournals.org/cgi/content/full/66/20/9852.
- ^ Ohgaki H, Eibl RH, Wiestler OD, Yasargil MG, Newcomb EW, Kleihues P (15 November 1991). "p53 mutations in nonastrocytic human brain tumors". Cancer Res. 51 (22): 6202–5. PMID 1933879. http://cancerres.aacrjournals.org/cgi/reprint/51/22/6202.
- ^ a b Ducray F, Idbaih A, de Reyniès A, et al. (2008). "Anaplastic oligodendrogliomas with 1p19q codeletion have a proneural gene expression profile". Mol. Cancer 7: 41. doi:10.1186/1476-4598-7-41. PMC 2415112. PMID 18492260. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2415112.
- ^ Ohgaki H, Kleihues P (June 2005). "Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas". J Neuropathol Exp Neurol. 64 (6): 479–89. PMID 15977639. http://meta.wkhealth.com/pt/pt-core/template-journal/lwwgateway/media/landingpage.htm?issn=0022-3069&volume=64&issue=6&spage=479.
- ^ a b c Hamlat A, Saikali S, Chaperon J, et al. (November 2005). "Oligodendroglioma: clinical study and survival analysis correlated with chromosomal anomalies". Neurosurg Focus 19 (5): E15. doi:10.3171/foc.2005.19.5.16. PMID 16398465. http://thejns.org/doi/pdf/10.3171/foc.2005.19.5.16.
- ^ Ino Y, Betensky RA, Zlatescu MC, et al. (1 April 2001). "Molecular subtypes of anaplastic oligodendroglioma: implications for patient management at diagnosis". Clin. Cancer Res. 7 (4): 839–45. PMID 11309331. http://clincancerres.aacrjournals.org/cgi/content/full/7/4/839.
- ^ Herbert H. Engelhard, M.D., Ph.D., Ana Stelea, M.D., and Arno Mundt, M.D., "Oligodendroglioma and Anaplastic Oligodendroglioma: Clinical Features, Treatment, and Prognosis",[1] p.452
- ^ See Temodar product information at http://www.spfiles.com/pitemodar.pdf
- ^ a b c A. B. Lassman, Oligodendroglioma Study Group; Memorial Sloan-Kettering Cancer Center, New York, NY (May 2009). "Retrospective analysis of outcomes among more than 1,000 patients with newly diagnosed anaplastic oligodendroglial tumors". J Clin Oncol, http://www.abstract.asco.org/AbstView_65_33109.html 27 (15s): suppl abstr 2014.
- ^ Kesari S, Schiff D, Drappatz J, et al. (January 2009). "Phase II Study of Protracted Daily Temozolomide for Low-Grade Gliomas in Adults". Clin. Cancer Res. 15 (1): 330–337. doi:10.1158/1078-0432.CCR-08-0888. PMID 19118062.
- ^ Coleman H, et al. (2002). "(Abstract) Impact of duration of temozolomide therapy on progression-free survival in recurrent malignant glioma". Neuro-Oncology 4 (4): 368. doi:10.1215/15228517-4-4-308. http://neuro-oncology.dukejournals.org/cgi/reprint/4/4/308.
- ^ Anderson K. and Lindsey W., "What is the optimal length of treatment with Temodar (temozolomide) for glioblastoma?", Online at http://virtualtrials.com/pdf/temodarlength.pdf
- ^ Sunyach MP, Jouvet A, Perol D, et al. (December 2007). "Role of exclusive chemotherapy as first line treatment in oligodendroglioma". J Neurooncol. 85 (3): 319–28. doi:10.1007/s11060-007-9422-3. PMID 17568995.
- ^ Mohile NA, Forsyth P, Stewart D, et al. (September 2008). "A phase II study of intensified chemotherapy alone as initial treatment for newly diagnosed anaplastic oligodendroglioma: an interim analysis". J Neurooncol. 89 (2): 187–93. doi:10.1007/s11060-008-9603-8. PMID 18458821.
- ^ Tatter SB (December 2002). "Recurrent malignant glioma in adults". Curr Treat Options Oncol 3 (6): 509–24. doi:10.1007/s11864-002-0070-8. PMID 12392640.
- ^ Herbert H. Engelhard, M.D., Ph.D., Ana Stelea, M.D., and Arno Mundt, M.D., "Oligodendroglioma and Anaplastic Oligodendroglioma: Clinical Features, Treatment, and Prognosis" [2] p.449
- ^ Feigenberg SJ, Amdur RJ, Morris CG, Mendenhall WM, Marcus RB, Friedman WA (2003). "Oligodendroglioma: does deferring treatment compromise outcome?". Am. J. Clin. Oncol. 26 (3): e60–6. PMID 12796617.
Nervous tissue tumors/NS neoplasm/Neuroectodermal tumor (ICD-O 9350–9589) (C70–C72, D32–D33, 191–192/225) Endocrine/
sellar (9350–9379)other: PinealomaCNS
(9380–9539)Astrocytoma (Pilocytic astrocytoma, Pleomorphic xanthoastrocytoma, Fibrillary (also diffuse or lowgrade) astrocytomas, Anaplastic astrocytoma, Glioblastoma multiforme)OligodendrogliomaEpendymoma · SubependymomaMultiple/unknownMature
neuronNeuroblastoma (Esthesioneuroblastoma, Ganglioneuroblastoma) · Medulloblastoma · Atypical teratoid rhabdoid tumorPrimitiveMeningiomas
(meninges)HematopoieticPNS: NST
(9540–9579)cranial and paraspinal nerves: Neurofibroma (Neurofibrosarcoma, Neurofibromatosis) · Neurilemmoma/Schwannoma (Acoustic neuroma) · Malignant peripheral nerve sheath tumornote: not all brain tumors are of nervous tissue, and not all nervous tissue tumors are in the brain (see brain metastases)
Categories:
Wikimedia Foundation. 2010.