- Congenital disorder of glycosylation
-
Congenital disorders of glycosylation Classification and external resources ICD-10 E77.8 ICD-9 271.8 OMIM 212065 212066 DiseasesDB 2012 31730 A congenital disorder of glycosylation (previously called carbohydrate-deficient glycoprotein syndrome) is one of several rare inborn errors of metabolism in which glycosylation of a variety of tissue proteins and/or lipids is deficient or defective. Congenital disorders of glycosylation are sometimes known as CDG syndromes. They often cause serious, sometimes fatal, malfunction of several different organ systems (especially the nervous system, muscles, and intestines) in affected infants. The most common subtype is CDG-Ia (also referred to as PMM2-CDG) where the genetic defect leads to the loss of phosphomannomutase 2, the enzyme responsible for the conversion of mannose-6-phosphate into mannose-1-phosphate.
Contents
History
The first CDG patients (twin sisters) were described in an abstract in the medical journal Pediatric Research in 1980 by Jaeken et al.[1] Their main features were psychomotor retardation, cerebral and cerebellar atrophy and fluctuating hormone levels (e.g.prolactin, FSH and GH). During the next 15 years the underlying defect remained unknown but since the plasmaprotein transferrin was underglycosylated (as shown by e.g. isoelectric focusing), the new syndrome was namned carbohydrate-deficient glycoprotein syndrome (CDGS).[2] Its "classical" phenotype included psychomotor retardation, ataxia, strabismus, anomalies (fat pads and inverted nipples) and coagulopathy.
In 1994, a new phenotype was described and namned CDGS-II.[3] In 1995, Van Schaftingen and Jaeken showed that CDGS-I (now CDG-Ia or PMM2-CDG) was caused by the deficiency of the enzyme phosphomannomutase. This enzyme is responsible for the interconversion of mannose-6-phosphate and mannose-1-phosphate, and its deficiency leads to a shortage in GDP-mannose and dolichol-mannose, two donors required for the synthesis of the lipid-linked oligosaccharide precursor of N-linked glycosylation.
In 1998, Niehues et al. published a new CDG syndrome, CDG-Ib, which is caused by mutations in the enzyme metabolically upstream of PMM2, phosphomannose isomerase (PMI).[4] In this paper, the authors also described a functional therapy for CDG-Ib, alimentary mannose.
The characterization of new defects took up speed and several new Type I and Type II defects were delineated.[5]
Classification
CDG can be classified as Types I and II (CDG-I and CDG-II), depending on the nature and location of the biochemical defect in the metabolic pathway relative to the action of oligosaccharyltransferase.
Currently, seventeen CDG type-I variants have been identified and twelve variants of CDG Type-II have been described.
Since 2009, most researchers use a different nomenclature based on the gene defect (e.g. CDG-Ia = PMM2-CDG, CDG-Ib = PMI-CDG, CDG-Ic = ALG6-CDG etc).[6] The reason for the new nomenclature was the fact that proteins not directly involved in glycan synthesis (such as members of the COG-family[7] and vesicular H+-ATPase [8]) were found to be causing the glycosylation defect in some CDG patients. Also, defects disturbing other glycosylation pathways than the N-linked one are included as CDG syndromes.
Type I
- Type I disorders involve disrupted synthesis of the lipid-linked oligosaccharide precursor.
Types include:
Type OMIM Gene Locus 1A 212065 PMM2 16p13.3-p13.2 1B 602579 MPI 15q22-qter 1C 603147 ALG6 1p22.3 1D 601110 ALG3 3q27 1E 608799 DPM1 20q13.13 1F 609180 MPDU1 17p13.1-p12 1G 607143 ALG12 22q13.33 1H 608104 ALG8 11pter-p15.5 1I 607906 ALG2 9q22 1J 608093 DPAGT1 11q23.3 1K 608540 ALG1 16p13.3 1L 608776 ALG9 11q23 1M 610768 TMEM15 9q34.11 1N 612015 RFT1 3p21.1 1O 612937 DPM3 1q12-q21 I/IIx 212067 n/a n/a Type II
- Type II disorders involve malfunctioning trimming/processing of the protein-bound oligosaccharide chain.
Types include:
Type OMIM Gene Locus 2A 212066 MGAT2 14q21 2B 606056 GCS1 2p13-p12 2C 266265 SLC35C1 11p11.2 2D 607091 B4GALT1 9p13 2E 608779 COG7 16p 2F 603585 SLC35A1 6q15 2G 611209 COG1 17q25.1 2H 611182 COG8 16q22.1 2I 613612 COG5 7q31 2J 613489 COG4 16q22.1 Presentation
The specific problems produced differ according to the particular abnormal synthesis involved. Common manifestations include ataxia; seizures; retinopathy; liver fibrosis; coagulopathies; failure to thrive; dysmorphic features (e.g., inverted nipples and subcutaneous fat pads; and strabismus. If an MRI is obtained, cerebellar atrophy and hypoplasia is a common finding.
Ocular abnormalities of CDG-Ia include: myopia, infantile esotropia, delayed visual maturation, low vision, optic pallor, and reduced rod function on electroretinography.[9]
Three subtypes of CDG I (a,b,d) can cause congenital hyperinsulinism with hyperinsulinemic hypoglycemia in infancy.[10]
Glycosylation and known defects
A biologically very important group of carbohydrates is the asparagine (Asn)-linked, or N-linked, oligosaccharides. Their biosynthetic pathway is very complex and involves a hundred or more glycosyltransferases, glycosidases, transporters[disambiguation needed
 ] and synthases. This plethora allows for the formation of a multitude of different final oligosaccharide structures, involved in protein folding, intracellular transport/localization, protein activity, and degradation/half-life. A vast amount of carbohydrate binding molecules (lectins) depend on correct glycosylation for appropriate binding; the selectins, involved in leukocyte extravasation, is a prime example. Their binding depends on a correct fucosylation of cell surface glycoproteins. Lack thereof leads to leukocytosis and increase sensitivity to infections as seen in SLC35C1-CDG(CDG-IIc); caused by a GDP-fucose (Fuc) transporter deficiency.
] and synthases. This plethora allows for the formation of a multitude of different final oligosaccharide structures, involved in protein folding, intracellular transport/localization, protein activity, and degradation/half-life. A vast amount of carbohydrate binding molecules (lectins) depend on correct glycosylation for appropriate binding; the selectins, involved in leukocyte extravasation, is a prime example. Their binding depends on a correct fucosylation of cell surface glycoproteins. Lack thereof leads to leukocytosis and increase sensitivity to infections as seen in SLC35C1-CDG(CDG-IIc); caused by a GDP-fucose (Fuc) transporter deficiency.All N-linked oligosaccharides originate from a common lipid-linked oligosaccharide (LLO) precursor, synthesized in the ER on a dolichol-phosphate (Dol-P) anchor. The mature LLO is transferred co-translationally to consensus sequence Asn residues in the nascent protein, and is further modified by trimming and re-building in the Golgi.
Deficiencies in the genes involved in N-linked glycosylation constitute the molecular background to most of the CDGs.
- Type I defects involve the synthesis and transfer of the LLO
- Type II defects impair the modification process of protein-bound oligosaccharides.
Type I
Description Disorder Product The formation of the LLO is initiated by the synthesis of the polyisoprenyl dolichol from farnesyl, a precursor of cholesterol biosynthesis. A recently published paper elegantly shows how an intermediary reductase in this process, is deficient in a Type I CDG (SRD5A3-CDG).[11] 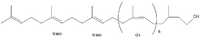
Dol is then activated to Dol-P via the action of Dol kinase in the ER membrane. This process is defective in DK1-CDG (CDG-Im).[12] 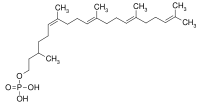
Consecutive N-acetylglucosamine (GlcNAc)- and mannosyltransferases use the nucleotide sugar donors UDP-GlcNAc and GDP-mannose (Man) to form a pyrophosphate-linked seven sugar glycan structure (Man5GlcNAc2-PP-Dol) on the cytoplasmatic side of the ER. Some of these steps have been found deficient in patients. - Deficiency in GlcNAc-1-P transferase causes DPAGT1-CDG (CDG-Ij)[13]
- Loss of the first mannosyltransferase causes ALG1-CDG (CDG-Ik)[14]
- Loss of the second mannosyltransferase causes ALG2-CDG (CDG-Ii).[15]
- Mutations in the other genes involved in these steps (ALG11, ALG13 and ALG14) are yet to be found.
Man5GlcNAc2-PP-Dol The M5GlcNAc2-structure is then flipped to the ER lumen, via the action of a "flippase" This is deficient in RFT1-CDG (CDG-In).[16] Finally, four mannosyltransferases and three glucosyltransferases complete the LLO structure Glc3Man9GlcNAc2-PP-Dol using Dol-P-Man and Dol-P-glucose (Glc) as donors. There are four known defects: - mannosyltransferase VI deficiency causes ALG3-CDG (CDG-Id)[17]
- mannosyltransferase VII/IX deficiency causes ALG9-CDG (CDG-IL)[18]
- mannosyltransferase VIII deficiency causes ALG12-CDG (CDG-Ig)[19]
- glucosyltransferase I deficiency causes ALG6-CDG (CDG-Ic)[20]
- glucosyltransferase II deficiency causes ALG8-CDG (CDG-Ih).[21]
Glc3Man9GlcNAc2-PP-Dol A protein with hitherto unknown activity, MPDU-1, is required for the efficient presentation of Dol-P-Man and Dol-P-Glc. Its deficiency causes MPDU1-CDG (CDG-If).[22] The synthesis of GDP-Man is crucial for proper N-glycosylation, as it serves as donor substrate for the formation of Dol-P-Man and the initial Man5GlcNAc2-P-Dol structure. GDP-Man synthesis is linked to glycolysis via the interconversion of fructose-6-P and Man-6-P, catalyzed by phosphomannose isomerase (PMI). This step is deficient in PMI-CDG (CDG-Ib),[23] which is the only treatable CDG-I subtype. 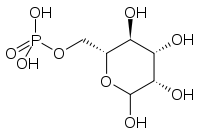
Man-1-P is then formed from Man-6-P, catalyzed by phosphomannomutase (PMM), and Man-1-P serves as substrate in the GDP-Man synthesis. Mutations in PMM2 cause PMM2-CDG (CDG-Ia), the most common CDG subtype. 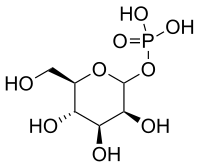
Dol-P-Man is formed via the action of Dol-P-Man synthase, consisting of three subunits; DPM1, DPM2, and DPM3. Mutations in DPM1 causes DPM1-CDG (CDG-Ie). No patients with mutations in DPM2 and DPM3 has been found so far. 
Type II
The mature LLO chain is next transferred to the growing protein chain, a process catalysed by the oligosaccharyl transferase (OST) complex.
- Once transferred to the protein chain, the oligosaccharide is trimmed by specific glycosidases. This process is vital since the lectin chaperones calnexin and calreticulin, involved in protein quality, bind to the Glc1Man9GlcNAc-structure and assure proper folding. Lack of the first glycosidase (GCS1) causes CDG-IIb.
- Removal of the Glc residues and the first Man residue occurs in the ER.
- The glycoprotein then travels to the Golgi, where a multitude of different structures with different biological activities are formed.
- Mannosidase I creates a Man5GlcNAc2-structure on the protein, but note that this has a different structure than the one made on LLO.
- Next, a GlcNAc residue forms GlcNAc1Man5GlcNAc2, the substrate for a-mannosidase II (aManII).
- aManII then removes two Man residues, creating the substrate for GlcNAc transferase II, which adds a GlcNAc to the second Man branch. This structure serves as substrate for additional galactosylation, fucosylation and sialylation reactions. Additionally, substitution with more GlcNAc residues can yield tri- and tetra-antennary molecules.
Not all structures are fully modified, some remain as high-mannose structures, others as hybrids (one unmodified Man branch and one modified), but the majority become fully modified complex type oligosaccharides.
In addition to glycosidase I, mutations have been found:
- in MGAT2, in GlcNAc transferase II (CDG-IIa)
- in SLC35C1, the GDP-Fuc transporter (CDG-IIc)
- in B4GALT1, a galactosyltransferase (CDG-IId)
- in COG7, the conserved oligomeric Golgi complex-7 (CDG-IIe)
- in SLC35A1, the CMP-sialic acid (NeuAc) transporter (CDG-IIf)
However, the use of >100 genes in this process, presumably means that many more defects are to be found.
Treatment
No treatment is available for most of these disorders. Mannose supplementation relieves the symptoms in PMI-CDG (CDG-Ib) for the most part,[24] even though the hepatic fibrosis may persist.[25] Fucose supplementation has had a partial effect on some SLC35C1-CDG (CDG-IIc or LAD-II) patients.[26]
See also
References
- ^ Jaeken, J., Vanderschueren-Lodeweyckx, M., Casaer, P., Snoeck, L., Corbeel, L., Eggermont, E., and Eeckels, R. (1980) Pediatr Res 14, 179
- ^ Jaeken, J., and Carchon, H. (1993) The carbohydrate-deficient glycoprotein syndromes: an overview. J Inherit Metab Dis. 16, 813-20.
- ^ Jaeken, J., Schachter, H., Carchon, H., De Cock, P., Coddeville, B. and Spik, G. (1994) Arch. Dis. Childhood 71, 123-127
- ^ Niehues, R., Hasilik, M., Alton, G., Körner, C., Schiebe-Sukumar, M., Koch, H.G., Zimmer, K.P., Wu, R., Harms, E., Reiter, K., von Figura, K., Freeze, H.H., Harms, H.K., Marquardt, T. Carbohydrate-deficient glycoprotein syndrome type Ib. Phosphomannose isomerase deficiency and mannose therapy. (1998) J. Clin. Invest. 101, 1414-20.
- ^ Haeuptle, M.A., and Hennet, T. Congenital disorders of glycosylation: an update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. (2009) Hum Mutat 30, 1628-41.
- ^ Jaeken, J., Hennet, T., Matthijs, G., and Freeze, H.H. (2009) CDG nomenclature: time for a change! Biochim Biophys Acta. 1792, 825-6.
- ^ Wu, X., Steet, R.A., Bohorov, O., Bakker, J., Newell, J., Krieger, M., Spaapen, L., Kornfeld, S., and Freeze, H.H. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. (2004) Nat. Med. 10, 518-23.
- ^ Kornak, U., Reynders, E., Dimopoulou, A., van Reeuwijk, J., Fischer, B., Rajab, A., Budde, B., Nürnberg, P., Foulquier, F.; ARCL Debré-type Study Group, Lefeber, D., Urban, Z., Gruenewald, S., Annaert, W., Brunner, H.G., van Bokhoven, H., Wevers, R., Morava, E., Matthijs, G., Van Maldergem, L., and Mundlos, S. Impaired glycosylation and cutis laxa caused by mutations in the vesicular H+-ATPase subunit ATP6V0A2. (2008) Nat. Genet. 40, 32-4.
- ^ Jensen H, Kjaergaard S, Klie F, Moller HU (2003). "Ophthalmic manifestations of congenital disorder of glycosylation type 1a". Ophthalmic Genet. 24 (2): 81–8. doi:10.1076/opge.24.2.81.13994. PMID 12789572.
- ^ Sun L, Eklund EA, Chung WK, Wang C, Cohen J, Freeze HH (2005). "Congenital disorder of glycosylation id presenting with hyperinsulinemic hypoglycemia and islet cell hyperplasia". J. Clin. Endocrinol. Metab. 90 (7): 4371–5. doi:10.1210/jc.2005-0250. PMID 15840742.
- ^ Cantagrel, V., Lefeber, D.J., Ng, B.G., Guan, Z., Silhavy, J.L., Bielas, S.L., Lehle, L., Hombauer, H., Adamowicz, M., Swiezewska, E., De Brouwer, A.P., Blümel, P., Sykut-Cegielska, .J, Houliston, S., Swistun, D., Ali, B.R., Dobyns, W.B., Babovic-Vuksanovic, D., van Bokhoven, H., Wevers, R.A., Raetz, C.R., Freeze, H.H., Morava, E., Al-Gazali, L., and Gleeson, J.G. SRD5A3 is required for converting polyprenol to dolichol and is mutated in a congenital glycosylation disorder. (2010) Cell, 142, 203-17
- ^ Kranz, C., Jungeblut, C., Denecke, J., Erlekotte, A., Sohlbach, C., Debus, V., Kehl ,H.G., Harms, E., Reith, A., Reichel, S., Grobe, H., Hammersen, G., Schwarzer, U., and Marquardt, T. A defect in dolichol phosphate biosynthesis causes a new inherited disorder with death in early infancy. (2007) Am. J. Hum. Genet., 80, 433-40.
- ^ Wu, X., Rush, J.S., Karaoglu, D., Krasnewich, D., Lubinsky, M.S., Waechter, C.J., Gilmore, R., and Freeze, H.H. Deficiency of UDP-GlcNAc:Dolichol Phosphate N-Acetylglucosamine-1 Phosphate Transferase (DPAGT1) causes a novel congenital disorder of Glycosylation Type Ij. (2003) Hum. Mut., 22, 144-50.
- ^ Grubenmann, C.E., Frank, C.G., Hülsmeier, A.J., Schollen, E., Matthijs, G., Mayatepek, E., Berger, E.G., Aebi, M., and Hennet, T. Deficiency of the first mannosylation step in the N-glycosylation pathway causes congenital disorder of glycosylation type Ik. (2004) Hum. Mol. Genet., 13-535-42.
- ^ Thiel, C., Schwarz, M., Peng, J., Grzmil, M., Hasilik, M., Braulke, T., Kohlschütter, A., von Figura, K., Lehle, L., and Körner, C. A new type of congenital disorders of glycosylation (CDG-Ii) provides new insights into the early steps of dolichol-linked oligosaccharide biosynthesis. (2003) J. Biol. Chem. 278, 22498-505.
- ^ Vleugels, W., Haeuptle, M.A., Ng, B.G., Michalski, J.C., Battini, R., Dionisi-Vici, C., Ludman, M.D., Jaeken, J., Foulquier, F., Freeze, H.H., Matthijs, G., and Hennet, T. RFT1 deficiency in three novel CDG patients. (2009) Hum. Mutat., 30, 1428-34.
- ^ Körner, C., Knauer, R., Stephani, U., Marquardt, T., Lehle, L.,and von Figura, K. Carbohydrate deficient glycoprotein syndrome type IV: deficiency of dolichyl-P-Man:Man(5)GlcNAc(2)-PP-dolichyl mannosyltransferase. (1999) EMBO J., 18, 6816-22.
- ^ Frank, C.G., Grubenmann, C.E., Eyaid, W., Berger, E.G., Aebi, M., and Hennet, T. Identification and functional analysis of a defect in the human ALG9 gene: definition of congenital disorder of glycosylation type IL. (2004) Am. J. Hum. Genet., 75, 146-50
- ^ Chantret, I., Dupré, T., Delenda, C., Bucher, S., Dancourt, J., Barnier, A., Charollais, A., Heron, D., Bader-Meunier, B., Danos, O., Seta, N., Durand, G., Oriol, R., Codogno, P., and Moore, S.E. Congenital disorders of glycosylation type Ig is defined by a deficiency in dolichyl-P-mannose:Man7GlcNAc2-PP-dolichyl mannosyltransferase. (2002) J. Biol. Chem., 277, 25815-22.
- ^ Körner, C., Knauer, R., Holzbach, U., Hanefeld, F., Lehle, L., and von Figura, K. Carbohydrate-deficient glycoprotein syndrome type V: deficiency of dolichyl-P-Glc:Man9GlcNAc2-PP-dolichyl glucosyltransferase. (1998) Proc. Natl. Acad. Sci U S A., 95, 13200-5.
- ^ Chantret, I., Dancourt, J., Dupré, T., Delenda, C., Bucher, S., Vuillaumier-Barrot, S., Ogier de Baulny, H., Peletan, C., Danos, O., Seta, N., Durand, G., Oriol, R., Codogno, P., and Moore, S.E. A deficiency in dolichyl-P-glucose:Glc1Man9GlcNAc2-PP-dolichyl alpha3-glucosyltransferase defines a new subtype of congenital disorders of glycosylation. (2003) J. Biol. Chem., 278, 9962-71.
- ^ Kranz, C., Denecke, J., Lehrman, M.A., Ray, S., Kienz, P., Kreissel, G., Sagi, D., Peter-Katalinic, J., Freeze, H.H., Schmid, T., Jackowski-Dohrmann, S., Harms, E., and Marquardt, T. A mutation in the human MPDU1 gene causes congenital disorder of glycosylation type If (CDG-If). (2001) J. Clin. Invest., 108, 1613-9.
- ^ Niehues R, Hasilik M, Alton G, Körner C, Schiebe-Sukumar M, Koch HG, Zimmer KP, Wu R, Harms E, Reiter K, von Figura K, Freeze HH, Harms HK, Marquardt T (1998). "Carbohydrate-deficient glycoprotein syndrome type Ib. Phosphomannose isomerase deficiency and mannose therapy". J. Clin. Invest. 101 (7): 1414–20. doi:10.1172/JCI2350. PMC 508719. PMID 9525984. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=508719.
- ^ Mention, K., Lacaille, F., Valayannopoulos, V., Romano, S., Kuster, A., Cretz, M., Zaidan, H., Galmiche, L., Jaubert, F., de Keyzer, Y., Seta, N., and de Lonlay, P. Development of liver disease despite mannose treatment in two patients with CDG-Ib. (2008) Mol. Genet. Metab. 93, 40-3
- ^ Westphal, V., Kjaergaard, S., Davis, J.A., Peterson, S.M., Skovby, F., and Freeze, H.H. Genetic and metabolic analysis of the first adult with congenital disorder of glycosylation type Ib: long-term outcome and effects of mannose supplementation. (2001) Mol. Genet. Metab. 73, 77-85.
- ^ Eklund, E.A., and Freeze, H.H. The congenital disorders of glycosylation: a multifaceted group of syndromes. (2006) NeuroRx 3, 254-63.
External links
- GeneReviews/NIH/NCBI/UW entry on PMM2-CDG (CDG-Ia)Carbohydrate-Deficient Glycoprotein Syndrome, Type 1a; Congenital Disorder of Glycosylation Type 1a; Jaeken Syndrome
- OMIM entries on Carbohydrate-Deficient Glycoprotein Syndrome, Type 1a; Congenital Disorder of Glycosylation Type 1a; Jaeken Syndrome
- The CDG Family Network
- GeneReviews/NIH/NCBI/UW entry on Congenital Disorders of Glycosylation Overview
Inborn error of carbohydrate metabolism: monosaccharide metabolism disorders (including glycogen storage diseases) (E73–E74, 271) Sucrose, transport
(extracellular)Disaccharide catabolismMonosaccharide transportHexose → glucose Monosaccharide catabolismGlucose ⇄ glycogen Glucose ⇄ CAC Pentose phosphate pathway Other (LSD) Inborn error of carbohydrate metabolism: glycoproteinosis (E77, 271.8) Anabolism Dolichol kinase deficiency · Congenital disorder of glycosylationPost-translational modification
of lysosomal enzymesCatabolism Aspartylglucosaminuria · Fucosidosis · mannosidosis (Alpha-mannosidosis, Beta-mannosidosis) · Sialidosis · Schindler diseaseOther Genetic disorder, membrane: Solute carrier disorders 1-10 SLC1A3 (Episodic ataxia 6) · SLC2A1 (De Vivo disease) · SLC2A5 (Fructose malabsorption) · SLC2A10 (Arterial tortuosity syndrome) · SLC3A1 (Cystinuria) · SLC4A1 (Hereditary spherocytosis 4/Hereditary elliptocytosis 4) · SLC4A11 (Congenital endothelial dystrophy type 2, Fuchs' dystrophy 4) · SLC5A1 (Glucose-galactose malabsorption) · SLC5A2 (Renal glycosuria) · SLC5A5 (Thyroid dyshormonogenesis type 1) · SLC6A19 (Hartnup disease) · SLC7A7 (Lysinuric protein intolerance) · SLC7A9 (Cystinuria)11-20 SLC11A1 (Crohn's disease) · SLC12A3 (Gitelman syndrome) · SLC16A1 (HHF7) · SLC16A2 (Allan–Herndon–Dudley syndrome) · SLC17A5 (Salla disease) · SLC17A8 (DFNA25)21-40 see also solute carrier family
B structural (perx, skel, cili, mito, nucl, sclr) · DNA/RNA/protein synthesis (drep, trfc, tscr, tltn) · membrane (icha, slcr, atpa, abct, othr) · transduction (iter, csrc, itra), trfkVesicle formation lysosome/melanosome: HPS1-HPS7 (Hermansky–Pudlak syndrome) · LYST (Chédiak–Higashi syndrome) ·
COPII: SEC23A (Cranio–lenticulo–sutural dysplasia)
COG7 (CDOG IIE)
APC: AP1S2 (X-Linked mental retardation 59) · AP3B1 (Hermansky–Pudlak syndrome 2) · AP4M1 (CPSQ3)Rab Cytoskeleton Vesicle fusion synaptic vesicle: SNAP29 (CEDNIK syndrome) · STX11 (Hemophagocytic lymphohistiocytosis 4)
caveolae: CAV1 (Congenital generalized lipodystrophy 3) · CAV3 (Limb-girdle muscular dystrophy 2B, Long QT syndrome 9)
vacuolar protein sorting: VPS33B (ARC syndrome) · VPS13B (Cohen syndrome)
DYSF (Distal muscular dystrophy)Categories:- Inborn errors of metabolism
- Pediatrics
- Glycoprotein metabolism disorders
- Membrane transport protein disorders




Wikimedia Foundation. 2010.