- Protein folding
-
"Protein thermodynamics" redirects here. For the thermodynamics of reactions catalyzed by proteins, see Enzyme.
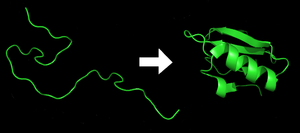 Protein before and after folding.
Protein before and after folding.
Protein folding is the process by which a protein structure assumes its functional shape or conformation. It is the physical process by which a polypeptide folds into its characteristic and functional three-dimensional structure from random coil.[1] Each protein exists as an unfolded polypeptide or random coil when translated from a sequence of mRNA to a linear chain of amino acids. This polypeptide lacks any developed three-dimensional structure (the left hand side of the neighboring figure). Amino acids interact with each other to produce a well-defined three-dimensional structure, the folded protein (the right hand side of the figure), known as the native state. The resulting three-dimensional structure is determined by the amino acid sequence (Anfinsen's dogma).[2]
The correct three-dimensional structure is essential to function, although some parts of functional proteins may remain unfolded[3] Failure to fold into native structure produces inactive proteins that are usually toxic. Several neurodegenerative and other diseases are believed to result from the accumulation of amyloid fibrills formed by misfolded proteins.[4] Many allergies are caused by the folding of the proteins, for the immune system does not produce antibodies for certain protein structures.[5]
Contents
Known facts
Relationship between folding and amino acid sequence
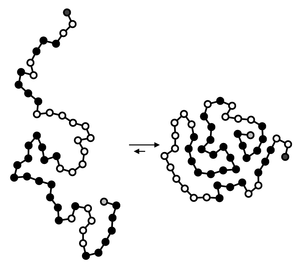 Illustration of the main driving force behind protein structure formation. In the compact fold (to the right), the hydrophobic amino acids (shown as black spheres) are in general shielded from the solvent.
Illustration of the main driving force behind protein structure formation. In the compact fold (to the right), the hydrophobic amino acids (shown as black spheres) are in general shielded from the solvent.The significant correlation between protein folding rates and the sequence-predicted secondary structure suggests that folding rates are largely determined by the amino acid sequence. The amino-acid sequence of a protein determines its native conformation.[6] A protein molecule folds spontaneously during or after biosynthesis. While these macromolecules may be regarded as "folding themselves", the process also depends on the solvent (water or lipid bilayer),[7] the concentration of salts, the temperature, and the presence of molecular chaperones.
Folded proteins usually have a side chain packing stabilizes the folded state, and charged or side chains occupy the solvent-exposed surface where they interact with surrounding water. Minimizing the number of hydrophobic side-chains exposed to water is an important driving force behind the folding process.[8] Formation of intramolecular hydrogen bonds provides another important contribution to protein stability.[9] The strength of hydrogen bonds depends on their environment, thus H-bonds enveloped in a hydrophobic core contribute more than H-bonds exposed to the aqueous environment to the stability of the native state.[10]
The process of folding often begins co-translationally, so that the N-terminus of the protein begins to fold while the C-terminal portion of the protein is still being synthesized by the ribosome. Specialized proteins called chaperones assist in the folding of other proteins.[11] A well studied example is the bacterial GroEL system, which assists in the folding of globular proteins. In eukaryotic organisms chaperones are known as heat shock proteins. Although most globular proteins are able to assume their native state unassisted, chaperone-assisted folding is often necessary in the crowded intracellular environment to prevent aggregation; chaperones are also used to prevent misfolding and aggregation which may occur as a consequence of exposure to heat or other changes in the cellular environment.
There are two models of protein folding that are currently being confirmed: The first: is the diffusion collision model in which a nucleus is formed, then the secondary structure, and finally these secondary structures are collided together and pack tightly together. The second: is the nucleation-condensation model, in which the secondary and tertiary structure of the protein is made at the same time. Finally, recent studies have shown that some proteins show characteristics of both of these folding models.
For the most part, scientists have been able to study many identical molecules folding together en masse. At the coarsest level, it appears that in transitioning to the native state, a given amino acid sequence takes on roughly the same route and proceeds through roughly the same intermediates and transition states. Often folding involves first the establishment of regular secondary and supersecondary structures, particularly alpha helices and beta sheets, and afterwards tertiary structure. Formation of quaternary structure usually involves the "assembly" or "coassembly" of subunits that have already folded. The regular alpha helix and beta sheet structures fold rapidly because they are stabilized by intramolecular hydrogen bonds, as was first characterized by Linus Pauling. Protein folding may involve covalent bonding in the form of disulfide bridges formed between two cysteine residues or the formation of metal clusters. Shortly before settling into their more energetically favourable native conformation, molecules may pass through an intermediate "molten globule" state.
The essential fact of folding, however, remains that the amino acid sequence of each protein contains the information that specifies both the native structure and the pathway to attain that state. This is not to say that nearly identical amino acid sequences always fold similarly.[12] Conformations differ based on environmental factors as well; similar proteins fold differently based on where they are found. Folding is a spontaneous process independent of energy inputs from nucleoside triphosphates. The passage of the folded state is mainly guided by hydrophobic interactions, formation of intramolecular hydrogen bonds, and van der Waals forces, and it is opposed by conformational entropy.
Disruption of the native state
Under some conditions proteins will not fold into their biochemically functional forms. Temperatures above or below the range that cells tend to live in will cause thermally unstable proteins to unfold or "denature" (this is why boiling makes an egg white turn opaque). High concentrations of solutes, extremes of pH, mechanical forces, and the presence of chemical denaturants can do the same. Protein thermal stability is far from constant, however. For example, hyperthermophilic bacteria have been found that grow at temperatures as high as 122 °C,[13] which of course requires that their full complement of vital proteins and protein assemblies be stable at that temperature or above.
A fully denatured protein lacks both tertiary and secondary structure, and exists as a so-called random coil. Under certain conditions some proteins can refold; however, in many cases denaturation is irreversible.[14] Cells sometimes protect their proteins against the denaturing influence of heat with enzymes known as chaperones or heat shock proteins, which assist other proteins both in folding and in remaining folded. Some proteins never fold in cells at all except with the assistance of chaperone molecules, which either isolate individual proteins so that their folding is not interrupted by interactions with other proteins or help to unfold misfolded proteins, giving them a second chance to refold properly. This function is crucial to prevent the risk of precipitation into insoluble amorphous aggregates.
Incorrect protein folding and neurodegenerative disease
Aggregated proteins are associated with prion-related illnesses such as Creutzfeldt-Jakob disease, bovine spongiform encephalopathy (mad cow disease), amyloid-related illnesses such as Alzheimer's disease and familial amyloid cardiomyopathy or polyneuropathy, as well as intracytoplasmic aggregation diseases such as Huntington's and Parkinson's disease.[4][15] These age onset degenerative diseases are associated with the multimerization of misfolded proteins into insoluble, extracellular aggregates and/or intracellular inclusions including cross-beta sheet amyloid fibrils; it is not clear whether the aggregates are the cause or merely a reflection of the loss of protein homeostasis, the balance between synthesis, folding, aggregation and protein turnover. Misfolding and excessive degradation instead of folding and function leads to a number of proteopathy diseases such as antitrypsin-associated emphysema, cystic fibrosis and the lysosomal storage diseases, where loss of function is the origin of the disorder. While protein replacement therapy has historically been used to correct the latter disorders, an emerging approach is to use pharmaceutical chaperones to fold mutated proteins to render them functional.
Effect of external factors on the folding of Proteins
Several external factors such as temperature, external fields (electric, magnetic),[16] molecular crowding,[17] limitation of space could have a big influence on the folding of proteins.[18] Modification of the local minima by external factors can also induce modifications of the folding trajectory.
Protein folding is a very finely tuned process. Hydrogen bonding between different atoms provides the force required. Hydrophobic interactions between hydrophobic amino acids pack the hydrophobic residues
The Levinthal paradox and kinetics
Levinthal's paradox is a thought experiment, also constituting a self-reference in the theory of protein folding. In 1969, Cyrus Levinthal noted that, because of the very large number of degrees of freedom in an unfolded polypeptide chain, the molecule has an astronomical number of possible conformations. An estimate of 3300 or 10143 was made in one of his papers.
The Levinthal paradox[19] observes that if a protein were folded by sequentially sampling of all possible conformations, it would take an astronomical amount of time to do so, even if the conformations were sampled at a rapid rate (on the nanosecond or picosecond scale). Based upon the observation that proteins fold much faster than this, Levinthal then proposed that a random conformational search does not occur, and the protein must, therefore, fold through a series of meta-stable intermediate states.
The duration of the folding process varies dramatically depending on the protein of interest. When studied outside the cell, the slowest folding proteins require many minutes or hours to fold primarily due to proline isomerization, and must pass through a number of intermediate states, like checkpoints, before the process is complete.[20] On the other hand, very small single-domain proteins with lengths of up to a hundred amino acids typically fold in a single step.[21] Time scales of milliseconds are the norm and the very fastest known protein folding reactions are complete within a few microseconds.[22]
Experimental techniques for studying protein folding
While inferences about protein folding can be made through mutation studies[citation needed]; typically, experimental techniques for studying protein folding rely on the gradual unfolding or folding of a solution of proteins and observing conformational changes using standard non-crystallographic techniques for observing protein structure.
Protein nuclear magnetic resonance spectroscopy
Main article: Protein NMRProtein folding is routinely studied using NMR spectroscopy, for example by monitoring hydrogen-deuterium exchange of partially folded intermediates.[citation needed]
Circular dichroism
Main article: Circular dichroismCircular dichroism is one of the most general and basic tools to study protein folding. Circular dichroism spectroscopy measures the absorption of circularly polarized light. In proteins, structures such as alpha helices and beta sheets are chiral, and thus absorb such light. The absorption of this light acts as a marker of the degree of foldedness of the protein ensemble. This technique has been used to measure equilibrium unfolding of the protein by measuring the change in this absorption as a function of denaturant concentration or temperature.[citation needed] A denaturant melt measures the free energy of unfolding as well as the protein's m value, or denaturant dependence. A temperature melt measures the melting temperature (Tm) of the protein. This type of spectroscopy can also be combined with fast-mixing devices, such as stopped flow, to measure protein folding kinetics and to generate chevron plots.
Dual polarisation interferometry
Main article: Dual polarisation interferometryDual polarisation interferometry is a surface based technique for measuring the optical properties of molecular layers. When used to characterise protein folding, it measures the conformation by determining the overall size of a monolayer of the protein and its density in real time at sub-Angstrom resolution[citation needed]. Although real time, measurement of the kinetics of protein folding are limited to processes that occur slower than ~10 Hz. Similar to circular dichroism the stimulus for folding can be a denaturant or temperature.
Vibrational circular dichroism of proteins
The more recent developments of vibrational circular dichroism (VCD) techniques for proteins, currently involving Fourier transform (FFT) instruments, provide powerful means for determining protein conformations in solution even for very large protein molecules. Such VCD studies of proteins are often combined with X-ray diffraction of protein crystals, FT-IR data for protein solutions in heavy water (D2O), or ab initio quantum computations to provide unambiguous structural assignments that are unobtainable from CD.[citation needed]
Studies of folding with high time resolution
The study of protein folding has been greatly advanced in recent years by the development of fast, time-resolved techniques. These are experimental methods for rapidly triggering the folding of a sample of unfolded protein, and then observing the resulting dynamics. Fast techniques in widespread use include neutron scattering,[23] ultrafast mixing of solutions, photochemical methods, and laser temperature jump spectroscopy. Among the many scientists who have contributed to the development of these techniques are Jeremy Cook, Heinrich Roder, Harry Gray, Martin Gruebele, Brian Dyer, William Eaton, Sheena Radford, Chris Dobson, Alan Fersht, Bengt Nölting and Lars Konermann.
Computational methods for studying protein folding
Main article: Protein structure predictionEnergy landscape of protein folding
The protein folding phenomenon was largely an experimental endeavor until the formulation of an energy landscape theory of proteins by Joseph Bryngelson and Peter Wolynes in the late 1980s and early 1990s. This approach introduced the principle of minimal frustration,.[24] This principle says that nature has chosen amino acid sequences so that the folded state of the protein is very stable. Additionally, the undesired interactions between amino acids along the folding pathway are reduced making the acquisition of the folded state a very fast process. Even though nature has reduced the level of frustration in proteins, some degree of it remains up to now as can be observed in the presence of local minima in the energy landscape of proteins. A consequence of these evolutionarily selected sequences is that proteins are generally thought to have globally "funneled energy landscapes" (coined by José Onuchic)[25] that are largely directed towards the native state. This "folding funnel" landscape allows the protein to fold to the native state through any of a large number of pathways and intermediates, rather than being restricted to a single mechanism. The theory is supported by both computational simulations of model proteins and experimental studies,[24] and it has been used to improve methods for protein structure prediction and design.[24] The description of protein folding by the leveling free-energy landscape is also consistent with the 2nd law of thermodynamics.[26] Physically, thinking of landscapes in terms of visualizable potential or total energy surfaces simply with maxima, saddle points, minima and funnels, rather like geographic landscapes, is perhaps a little misleading. The relevant description is really a highly dimensional phase space in which manifolds might take a variety of more complicated topological forms.[27]
Modeling of protein folding
De novo or ab initio techniques for computational protein structure prediction are related to, but strictly distinct from experimental studies of protein folding. Molecular Dynamics (MD) is an important tool for studying protein folding and dynamics in silico. First equilibrium folding simulations were done using implicit solvent model and Umbrella Sampling.[28] Because of computational cost, ab initio MD folding simulations with explicit water are limited to peptides and very small proteins.[29][30] MD simulations of larger proteins remain restricted to dynamics of the experimental structure or its high-temperature unfolding. In order to simulate long time folding processes (beyond about 1 microsecond), like folding of small-size proteins (about 50 residues) or larger, some approximations or simplifications in protein models need to be introduced. An approach using reduced protein representation (pseudo-atoms representing groups of atoms are defined) and statistical potential are useful in protein structure prediction and modeling of the folding pathways.[31]
There are distributed computing projects which use idle CPU or GPU time of personal computers to solve problems such as protein folding or prediction of protein structure, one prominent example being the Folding@Home project. People can run these programs on their computer or PlayStation 3 to support them.
See also
- Chevron plot
- Denaturation (biochemistry)
- Denaturation midpoint
- Downhill folding
- Folding (chemistry)
- Folding@Home
- Foldit computer game
- Levinthal paradox
- Protein design
- Protein dynamics
- Protein Misfolding Cyclic Amplification
- Protein structure prediction
- Protein structure prediction software
- Proteopathy
- Rosetta@home
- Statistical potential
- Software for molecular mechanics modeling
References
- ^ Alberts, Bruce; Alexander Johnson, Julian Lewis, Martin Raff, Keith Roberts, and Peter Walters (2002). "The Shape and Structure of Proteins". Molecular Biology of the Cell; Fourth Edition. New York and London: Garland Science. ISBN 0-8153-3218-1. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Search&db=books&doptcmdl=GenBookHL&term=mboc4%5Bbook%5D+AND+372270%5Buid%5D&rid=mboc4.section.388.
- ^ Anfinsen, C. (1972). "The formation and stabilization of protein structure". Biochem. J. 128 (4): 737–49. PMC 1173893. PMID 4565129. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1173893.
- ^ Jeremy M. Berg, John L. Tymoczko, Lubert Stryer; Web content by Neil D. Clarke (2002). "3. Protein Structure and Function". Biochemistry. San Francisco: W. H. Freeman. ISBN 0-7167-4684-0. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Search&db=books&doptcmdl=GenBookHL&term=stryer%5Bbook%5D+AND+215168%5Buid%5D&rid=stryer.chapter.280.
- ^ a b Dennis J. Selkoe (2003). "Folding proteins in fatal ways". Nature 426 (6968): 900–904. doi:10.1038/nature02264. PMID 14685251. http://www.nature.com/nature/journal/v426/n6968/full/nature02264.html.
- ^ Alberts, Bruce, Dennis Bray, Karen Hopkin, Alexander Johnson, Julian Lewis, Martin Raff, Keith Roberts, and Peter Walter. "Protein Structure and Function." Essential Cell Biology. Edition 3. New York: Garland Science, Taylor and Francis Group, LLC, 2010. Pg 120-170.
- ^ Anfinsen CB. (20 July 1973). "Principles that Govern the Folding of Protein Chains". Science. 181 (4096): 223–230. doi:10.1126/science.181.4096.223. PMID 4124164. http://www.sciencemag.org/cgi/pdf_extract/181/4096/223.
- ^ van den Berg, B., Wain, R., Dobson, C. M., Ellis R. J. (August 2000). "Macromolecular crowding perturbs protein refolding kinetics: implications for folding inside the cell". EMBO J. 19 (15): 3870–5. doi:10.1093/emboj/19.15.3870. PMC 306593. PMID 10921869. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=306593.
- ^ Pace, C., Shirley, B., McNutt, M., Gajiwala, K. (1 January 1996). "Forces contributing to the conformational stability of proteins". FASEB J. 10 (1): 75–83. PMID 8566551. http://www.fasebj.org/cgi/reprint/10/1/75.
- ^ Rose, G., Fleming, P., Banavar, J., Maritan, A. (2006). "A backbone-based theory of protein folding". Proc. Natl. Acad. Sci. U.S.A. 103 (45): 16623–33. doi:10.1073/pnas.0606843103. PMC 1636505. PMID 17075053. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1636505.
- ^ Deechongkit, S., Nguyen, H., Dawson, P. E., Gruebele, M., Kelly, J. W. (2004). "Context Dependent Contributions of Backbone H-Bonding to β-Sheet Folding Energetics". Nature 403 (45): 101–5. doi:10.1038/nature02611. PMID 15229605.
- ^ Lee, S., Tsai, F. (2005). "Molecular chaperones in protein quality control". J. Biochem. Mol. Biol. 38 (3): 259–65. doi:10.5483/BMBRep.2005.38.3.259. PMID 15943899. http://www.jbmb.or.kr/fulltext/jbmb/view.php?vol=38&page=259.
- ^ Alexander, P. A., He Y., Chen, Y., Orban, J., Bryan, P. N. (2007). "The design and characterization of two proteins with 88% sequence identity but different structure and function". Proc Natl Acad Sci U S A. 104 (29): 11963–8. doi:10.1073/pnas.0700922104. PMC 1906725. PMID 17609385. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1906725.
- ^ Takai, K., Nakamura, K., Toki, T., Tsunogai, U., Miyazaki, M., Miyazaki, J., Hirayama, H., Nakagawa, S., Nunoura, T., Horikoshi, K. (2008). "Cell proliferation at 122 °C and isotopically heavy CH4 production by a hyperthermophilic methanogen under high-pressure cultivation". Proc Natl Acad Sci USA 105 (31): 10949–54. doi:10.1073/pnas.0712334105. PMC 2490668. PMID 18664583. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2490668.
- ^ Shortle, D. (1 January 1996). "The denatured state (the other half of the folding equation) and its role in protein stability". FASEB J. 10 (1): 27–34. PMID 8566543. http://www.fasebj.org/cgi/reprint/10/1/27.
- ^ Chiti, F.; Dobson, C. (2006). "Protein misfolding, functional amyloid, and human disease.". Annual review of biochemistry 75: 333–366. doi:10.1146/annurev.biochem.75.101304.123901. PMID 16756495.
- ^ Ojeda, P., Garcia, M. (2010). "Electric Field-Driven Disruption of a Native β-Sheet Protein Conformation and Generation of a Helix-Structure". Biophysical Journal 99 (2): 595–599. Bibcode 2010BpJ....99..595O. doi:10.1016/j.bpj.2010.04.040. PMC 2905109. PMID 20643079. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2905109.
- ^ Berg, B., Ellis, J., Dobson, C. (1999). "Effects of macromolecular crowding on protein folding and aggregation". The EMBO Journal 18 (24): 6927–6933. doi:10.1093/emboj/18.24.6927. PMC 1171756. PMID 10601015. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1171756.
- ^ Ellis RJ (July 2006). "Molecular chaperones: assisting assembly in addition to folding". Trends in Biochemical Sciences 31 (7): 395–401. doi:10.1016/j.tibs.2006.05.001. PMID 16716593.
- ^ C. Levinthal (1968). "Are there pathways for protein folding?". J. Chim. Phys. 65: 44–5. http://www.biochem.wisc.edu/courses/biochem704/Reading/Levinthal1968.pdf.
- ^ Kim, P. S., Baldwin, R. L. (1990). "Intermediates in the folding reactions of small proteins". Annu. Rev. Biochem. 59: 631–60. doi:10.1146/annurev.bi.59.070190.003215. PMID 2197986.
- ^ Jackson S. E. (August 1998). "How do small single-domain proteins fold?". Fold Des 3 (4): R81–91. doi:10.1016/S1359-0278(98)00033-9. PMID 9710577. http://biomednet.com/elecref/13590278003R0081.[dead link]
- ^ Kubelka, J., Hofrichter, J., Eaton, W. A. (February 2004). "The protein folding 'speed limit'". Curr. Opin. Struct. Biol. 14 (1): 76–88. doi:10.1016/j.sbi.2004.01.013. PMID 15102453.
- ^ Bu, Z; Cook, J; Callaway, D. J. E. (2001). "Dynamic regimes and correlated structural dynamics in native and denatured alpha-lactalbuminC". J Mol Biol 312 (4): 865–873. doi:10.1006/jmbi.2001.5006. PMID 11575938.
- ^ a b c Bryngelson, J. D., Onuchic, J. N., Socci, N. D. and Wolynes, P.G. (1995). "Funnels, Pathways, and the Energy Landscape of Protein Folding: A Synthesis". Proteins:Struct. Funct. Genet. 21 (3): 167–195. doi:10.1002/prot.340210302. PMID 7784423. http://wolynes.ucsd.edu/Wolynes%20Papers/Funnels%20Pathways%20135.pdf.
- ^ Leopold, P. E., Montal, M. and Onuchic, J. N. (1992). "Protein folding funnels: a kinetic approach to the sequence-structure relationship". Proc. Natl. Acad. Sci. USA 89 (18): 8721–8725. doi:10.1073/pnas.89.18.8721. PMC 49992. PMID 1528885. http://www.pnas.org/content/89/18/8721.full.pdf+html.
- ^ Sharma, V., Kaila, V. R. I., and Annila, A. (2009). "Protein folding as an evolutionary process". Physica A 388 (6): 851–862. doi:10.1016/j.physa.2008.12.004.
- ^ Robson, B, Vaithilingham A. (2008). Protein Folding Revisited. Progress in Molecular Biology and Translational Science, Molecular Biology of Protein Folding. 84:161-202, Elsevier Press/Academic Press
- ^ Schaefer, Michael; Bartels, Christian, Karplus, Martin (3 December 1998). "Solution conformations and thermodynamics of structured peptides: molecular dynamics simulation with an implicit solvation model1". Journal of Molecular Biology 284 (3): 835–848. doi:10.1006/jmbi.1998.2172. PMID 9826519.
- ^ "Fragment-based Protein Folding Simulations". http://www.cs.ucl.ac.uk/staff/d.jones/t42morph.html.
- ^ "Protein folding" (by Molecular Dynamics). http://www.biomolecular-modeling.com/Abalone/Protein-folding.html.
- ^ Kmiecik, S., and Kolinski, A. (2007). "Characterization of protein-folding pathways by reduced-space modeling". Proc. Natl. Acad. Sci. U.S.A. 104 (30): 12330–5. doi:10.1073/pnas.0702265104. PMC 1941469. PMID 17636132. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1941469.
External links
- FoldIt - Folding Protein Game
- Folding@Home
- Rosetta@Home
- Human Proteome Folding Project
- BHAGEERATH-H: Protein tertiary structure prediction server
General All-α folds: All-β folds: α/β folds: α+β folds: Irregular folds: ←Secondary structureProcesses Protein biosynthesis · Posttranslational modification · Protein folding · Protein targeting · Proteome · Protein methodsStructures Types List of types of proteins · List of proteins · Membrane protein · Globular protein (Globulin, Albumin) · Fibrous proteinbiochemical families: prot · nucl · carb (glpr, alco, glys) · lipd (fata/i, phld, strd, gllp, eico) · amac/i · ncbs/i · ttpy/i
B proteins: BY STRUCTURE: membrane, globular (en, ca, an), fibrousProtein structure Nucleic acid structure See also Chaperones/
protein foldingOtherProtein targeting Ubiquitin E1 Ubiquitin-activating enzyme (UBA1, UBA2, UBA3, UBA5, UBA6, UBA7, ATG7, NAE1, SAE1)
E2 Ubiquitin-conjugating enzyme (A • B • C • D1, D2, D3 • E1, E2, E3 • G1, G2 • H • I • J1, J2 • K • L1, L2, L3, L4, L6 • M • N • O • Q1, Q2 • R1 (CDC34), R2 • S • V1, V2 • Z)
E3 Ubiquitin ligase (VHL, Cullin, CBL, MDM2, FANCL, UBR1)
Deubiquitinating enzyme: Ataxin 3 • USP6 • CYLD
ATG3 • BIRC6 • UFC1Other Categories:
Wikimedia Foundation. 2010.