- Intrinsically unstructured proteins
-
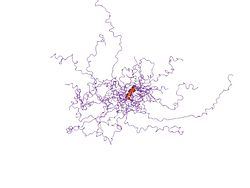 An ensemble of NMR structures of the Thylakoid soluble phosphoprotein TSP9, which shows a largely flexible protein chain.[1]
An ensemble of NMR structures of the Thylakoid soluble phosphoprotein TSP9, which shows a largely flexible protein chain.[1]
Intrinsically unstructured proteins, often referred to as naturally unfolded proteins or disordered proteins, are proteins characterized by lack of stable tertiary structure when the protein exists as an isolated polypeptide chain (a subunit) under physiological conditions in vitro.[2][3] The discovery of intrinsically unfolded proteins challenged the traditional protein structure paradigm, which states that a specific well-defined structure was required for the correct function of a protein and that the structure defines the function of the protein. This is clearly not the case for intrinsically unfolded proteins that remain functional despite the lack of a well-defined structure. Such proteins, in some cases, can adopt a fixed three dimensional structure after binding to other macromolecules.
Contents
Biological role of intrinsic disorder
Many disordered proteins have the binding affinity with their receptors regulated by post-translational modification, thus it has been proposed that the flexibility of disordered proteins facilitates the different conformational requirements for binding the modifying enzymes as well as their receptors.[4] Intrinsic disorder is particularly enriched in proteins implicated in cell signaling, transcription and chromatin remodeling functions.[5][6]
Flexible linkers
Disordered regions are often found as flexible linkers (or loops) connecting two globular or transmembrane domains. Linker sequences vary greatly in length and amino acid sequence, but are similar in amino acid composition (rich in polar uncharged amino acids). Flexible linkers allow the connecting domains to freely twist and rotate through space to recruit their binding partners or for those binding partners to induce larger scale interdomain conformation changes.
Coupled folding and binding
Many unstructured proteins undergo transitions to more ordered states upon binding to their targets. The coupled folding and binding may be local, involving only a few interacting residues, or it might involve an entire protein domain. It was recently shown that the coupled folding and binding allows the burial of a large surface area that would be possible only for fully structured proteins if they were much larger.[7] Moreover, certain disordered regions might serve as "molecular switches" in regulating certain biological function by switching to ordered conformation upon molecular recognition like small molecule-binding, DNA/RNA binding, ion interactions etc.[8].
The ability of disordered proteins to bind, and thus to exert a function, shows that stability is not a required condition. Many short functional sites, for example Short Linear Motifs are over-represented in disordered proteins.
Sequence signatures of disorder
Intrinsically unstructured proteins are characterized by a low content of bulky hydrophobic amino acids and a high proportion of polar and charged amino acids. Thus disordered sequences cannot bury sufficient hydrophobic core to fold like stable globular proteins. In some cases, hydrophobic clusters in disordered sequences provide the clues for identifying the regions that undergo coupled folding and binding. Such signatures are the basis of the prediction methods below.
Many disordered proteins also reveal low complexity sequences, i.e. sequences with overrepresentation of a few residues. While low complexity sequences are a strong indication of disorder, the reverse is not necessarily true, that is, not all disordered proteins have low complexity sequences. Disordered proteins have a low content of predicted secondary structure.
Identification of intrinsically unstructured proteins
Intrinsically unfolded proteins, once purified, can be identified by various experimental methods. Folded proteins have a high density (partial specific volume of 0.72-0.74 mL/g) and commensurately small radius of gyration. Hence, unfolded proteins can be detected by methods that are sensitive to molecular size, density or hydrodynamic drag, such as size exclusion chromatography, analytical ultracentrifugation, Small angle X-ray scattering (SAXS), and measurements of the diffusion constant. Unfolded proteins are also characterized by their lack of secondary structure, as assessed by far-UV (170-250 nm) circular dichroism (esp. a pronounced minimum at ~200 nm) or infrared spectroscopy.
Unfolded proteins have exposed backbone peptide groups exposed to solvent, so that they are readily cleaved by proteases, undergo rapid hydrogen-deuterium exchange and exhibit a small dispersion (<1 ppm) in their 1H amide chemical shifts as measured by NMR. (Folded proteins typically show dispersions as large as 5 ppm for the amide protons.)
The primary method to obtain information on disordered regions of a protein is NMR spectroscopy. The lack of electron density in X-ray crystallographic studies may also be a sign of disorder.
Disorder and Disease
Intrinsically unstructured proteins have been implicated in a number of diseases. Aggregation of misfolded proteins is the cause of many synucleinopathies. The aggregation of the intrinsically unstructured protein α-Synuclein is thought to be responsible. The structural flexibility of this protein together with its susceptibility to modification in the cell leads to misfolded and aggregation.
Many key oncogenes have large intrinsically unstructured regions, for example p53 and BRCA1. These regions of the proteins are responsible for mediating many of their interactions.
De novo prediction of intrinsically unstructured proteins
Computational methods exploit the sequence signatures of disorder to predict whether a protein is disordered given its amino acid sequence. The table below, which was originally adapted from[9] and has been recently updated, shows the main features of software for disorder prediction. Note that different software use different definitions of disorder.
Predictor What is predicted Based on Generates and uses multiple sequence alignment? PONDR All regions that are not rigid including random coils, partially unstructured regions, and molten globules Local aa composition, flexibility, hydropathy, etc No GlobPlot Regions with high propensity for globularity on the Russell/Linding scale (propensities for secondary structures and random coils) Russell/Linding scale of disorder No DisEMBL LOOPS (regions devoid of regular secondary structure); HOT LOOPS (highly mobile loops); REMARK465 (regions lacking electron density in crystal structure) Neural networks trained on X-ray structure data No SEG Low-complexity segments that is, “simple sequences” or “compositionally biased regions”. Locally optimized low-complexity segments are produced at defined levels of stringency and then refined according to the equations of Wootton and Federhen No Disopred2[10] Regions devoid of ordered regular secondary structure Cascaded support vector machine classifiers trained on PSI-BLAST profiles Yes OnD-CRF[11] The transition between structurally ordered and mobile or disordered amino acids intervals under native conditions. OnD-CRF applies Conditional Random Fields, CRFs, which rely on features generated from the amino acid sequence and from secondary structure prediction. No NORSp Regions with No Ordered Regular Secondary Structure (NORS). Most, but not all, are highly flexible. Secondary structure and solvent accessibility Yes FoldIndex[12] Regions that have a low hydrophobicity and high net charge (either loops or unstructured regions) Charge/hydrophaty analyzed locally using a sliding window No Charge/hydropathy method.[13] Fully unstructured domains (random coils) Global sequence composition No HCA (Hydrophobic Cluster Analysis) Hydrophobic clusters, which tend to form secondary structure elements Helical visualization of amino acid sequence No PreLink Regions that are expected to be unstructured in all conditions, regardless of the presence of a binding partner Compositional bias and low hydrophobic cluster content. No IUPred Regions that lack a well-defined 3D-structure under native conditions Energy resulting from inter-residue interactions, estimated from local amino acid composition No RONN Regions that lack a well-defined 3D structure under native conditions Bio-basis function neural network trained on disordered proteins No MD (Meta-Disorder predictor) Regions of different "types"; for example, unstructured loops and regions containing few stable intra-chain contacts A neural-network based meta-predictor that uses different sources of information predominantly obtained from orthogonal approaches Yes GeneSilico Metadisorder[14] Regions that lack a well-defined 3D structure under native conditions (REMARK-465) Meta method, which uses other disorder predictors (like RONN, IUPred, POODLE, and many more). Based on them the consensus is calculated according method accuracy (optimized using ANN, filtering and other techniques). Currently the best available method (first 2 places in last CASP experiment (blind test)) Yes IUPforest-L Long disordered regions in a set of proteins Moreau-Broto auto-correlation function of amino acid indices (AAIs) No MFDp [15] Different types of disorder including random coils, unstructured regions, molten globules, and REMARK-465-based regions. An ensemble of 3 SVMs specialized for the prediction of short, long and generic disordered regions, which combines three complementary disorder predictors, sequence, sequence profiles, predicted secondary structure, solvent accessibility, backbone dihedral torsion angles, residue flexibility and B-factors. MFDp (unofficially) secured 3rd place in last CASP experiment) Yes Since the methods above use different definitions of disorder and they were trained on different datasets, it is difficult to estimate their relative accuracy, but disorder prediction category is a part of biannual CASP experiment that is designed to test methods according accuracy in finding regions with missing 3D structure.
See also
References
- ^ Song J, Lee MS, Carlberg I, Vener AV, Markley JL (December 2006). "Micelle-induced folding of spinach thylakoid soluble phosphoprotein of 9 kDa and its functional implications". Biochemistry 45 (51): 15633–43. doi:10.1021/bi062148m. PMC 2533273. PMID 17176085. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2533273.
- ^ Dyson HJ, Wright PE (March 2005). "Intrinsically unstructured proteins and their functions". Nat. Rev. Mol. Cell Biol. 6 (3): 197–208. doi:10.1038/nrm1589. PMID 15738986.
- ^ Dunker AK, Silman I, Uversky VN, Sussman JL (December 2008). "Function and structure of inherently disordered proteins". Curr. Opin. Struct. Biol. 18 (6): 756–64. doi:10.1016/j.sbi.2008.10.002. PMID 18952168. http://linkinghub.elsevier.com/retrieve/pii/S0959-440X(08)00151-6.
- ^ Collins MO, Yu L, Campuzano I, Grant SG, Choudhary JS (July 2008). "Phosphoproteomic analysis of the mouse brain cytosol reveals a predominance of protein phosphorylation in regions of intrinsic sequence disorder". Mol. Cell Proteomics 7 (7): 1331–48. doi:10.1074/mcp.M700564-MCP200. PMID 18388127. http://www.mcponline.org/cgi/pmidlookup?view=long&pmid=18388127.
- ^ Iakoucheva LM, Brown CJ, Lawson JD, Obradović Z, Dunker AK (October 2002). "Intrinsic disorder in cell-signaling and cancer-associated proteins". J. Mol. Biol. 323 (3): 573–84. doi:10.1016/S0022-2836(02)00969-5. PMID 12381310. http://linkinghub.elsevier.com/retrieve/pii/S0022283602009695.
- ^ Sandhu KS (2009). "Intrinsic disorder explains diverse nuclear roles of chromatin remodeling proteins". J. Mol. Recognit. 22 (1): 1–8. doi:10.1002/jmr.915. PMID 18802931.
- ^ Gunasekaran K, Tsai CJ, Kumar S, Zanuy D, Nussinov R (February 2003). "Extended disordered proteins: targeting function with less scaffold". Trends Biochem. Sci. 28 (2): 81–5. doi:10.1016/S0968-0004(03)00003-3. PMID 12575995. http://linkinghub.elsevier.com/retrieve/pii/S0968000403000033.
- ^ Sandhu KS, Dash D (July 2007). "Dynamic alpha-helices: conformations that do not conform". Proteins 68 (1): 109–22. doi:10.1002/prot.21328. PMID 17407165.
- ^ Ferron F, Longhi S, Canard B, Karlin D (October 2006). "A practical overview of protein disorder prediction methods". Proteins 65 (1): 1–14. doi:10.1002/prot.21075. PMID 16856179.
- ^ Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT (March 2004). "Prediction and functional analysis of native disorder in proteins from the three kingdoms of life". J. Mol. Biol. 337 (3): 635–45. doi:10.1016/j.jmb.2004.02.002. PMID 15019783. http://linkinghub.elsevier.com/retrieve/pii/S0022283604001482.
- ^ Wang L, Sauer UH (June 2008). "OnD-CRF: predicting order and disorder in proteins using conditional random fields". Bioinformatics 24 (11): 1401–2. doi:10.1093/bioinformatics/btn132. PMID 18430742. http://bioinformatics.oxfordjournals.org/cgi/pmidlookup?view=long&pmid=18430742.
- ^ Prilusky J, Felder CE, Zeev-Ben-Mordehai T, et al. (August 2005). "FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded". Bioinformatics 21 (16): 3435–8. doi:10.1093/bioinformatics/bti537. PMID 15955783. http://bioinformatics.oxfordjournals.org/cgi/pmidlookup?view=long&pmid=15955783.
- ^ Uversky VN, Gillespie JR, Fink AL (November 2000). "Why are "natively unfolded" proteins unstructured under physiologic conditions?". Proteins 41 (3): 415–27. doi:10.1002/1097-0134(20001115)41:3<415::AID-PROT130>3.0.CO;2-7. PMID 11025552.
- ^ Schlessinger A, Punta M, Yachdav G, Kajan L, Rost B (2009). Orgel, Joseph P. R.14 O.. ed. "Improved disorder prediction by combination of orthogonal approaches". PLoS ONE 4 (2): e4433. doi:10.1371/journal.pone.0004433. PMC 2635965. PMID 19209228. http://dx.plos.org/10.1371/journal.pone.0004433.
- ^ Mizianty MJ, Stach W, Chen K, Kedarisetti KD, Disfani FM, Kurgan L (September 2010). "Improved sequence-based prediction of disordered regions with multilayer fusion of multiple information sources". Bioinformatics 26 (18): i489–96. doi:10.1093/bioinformatics/btq373. PMC 2935446. PMID 20823312. http://bioinformatics.oxfordjournals.org/cgi/pmidlookup?view=long&pmid=20823312.
Categories:- Proteins
- Protein structure
- Proteomics
Wikimedia Foundation. 2010.