- Lysinuric protein intolerance
-
Lysinuric protein intolerance Classification and external resources 
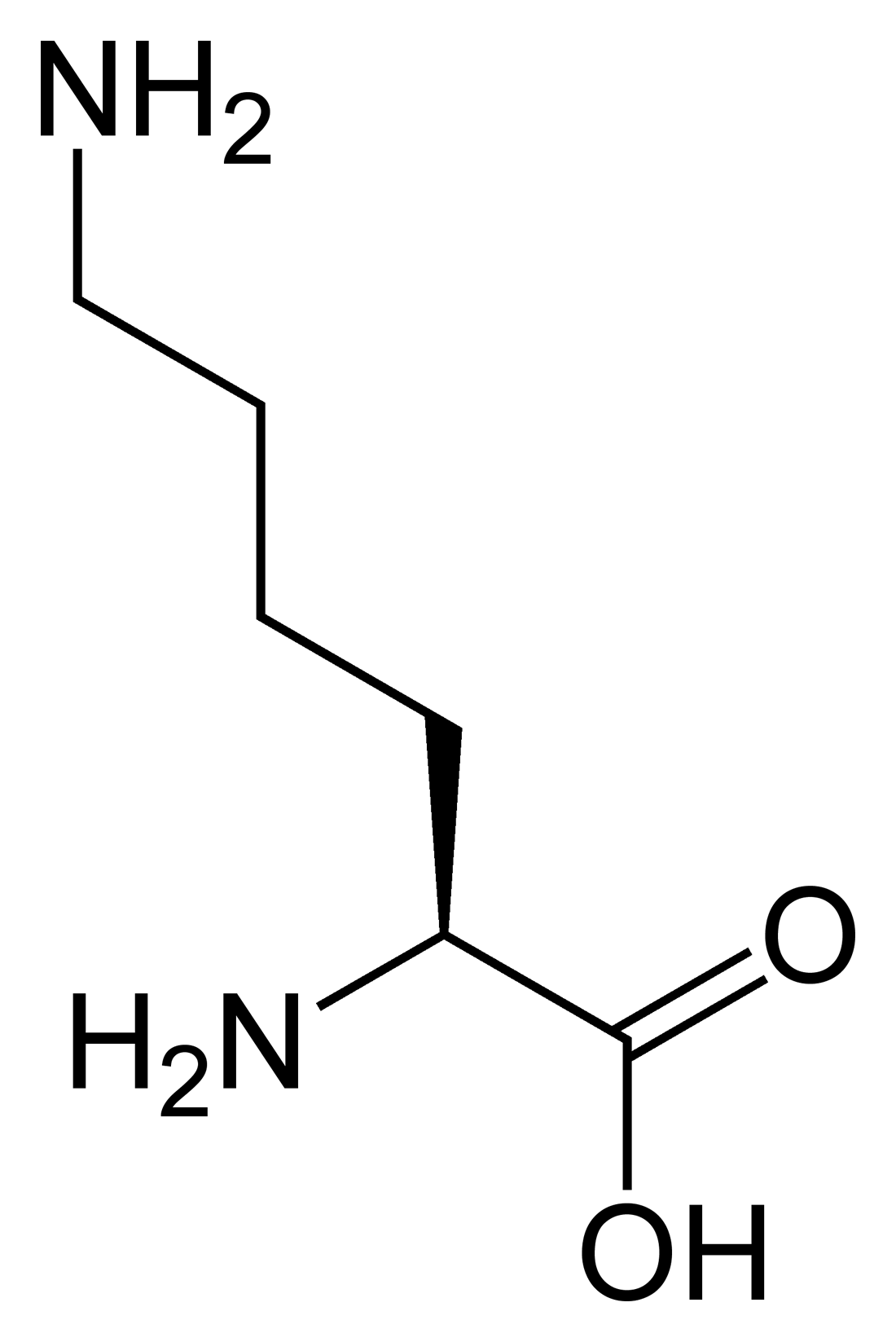
LysineICD-10 E72.3 OMIM 222700 DiseasesDB 29819 Lysinuric protein intolerance (LPI), also called hyperdibasic aminoaciduria type 2 or familial protein intolerance, is an autosomal recessive[1] metabolic disorder affecting amino acid transport.
About 100 patients have been reported, almost half of them of Finnish origin.
Contents
Causes
It has been associated with SLC7A7.[2]
Presentation
 Lysinuric protein intolerance has an autosomal recessive pattern of inheritance.
Lysinuric protein intolerance has an autosomal recessive pattern of inheritance.
In LPI, urinary excretion of cationic amino acids (ornithine, arginine and lysine) is increased and these amino acids are poorly absorbed from the intestine. Therefore, their plasma concentrations are low and their body pools become depleted. Deficiency of arginine and ornithine restricts the function of the urea cycle and leads to hyperammonemia after protein-rich meals. Deficiency of lysine may play a major role in the skeletal and immunological abnormalities observed in LPI patients.
Diagnosis
The diagnosis is based on the biochemical findings (increased concentrations of lysine, arginine and ornithine in urine and low concentrations of these amino acids in plasma, elevation of urinary orotic acid excretion after protein-rich meals, and inappropriately high concentrations of serum ferritin and lactate dehydrogenase isoenzymes) and the screening of known mutations of the causative gene from a DNA sample.
Symptoms
Infants with LPI are usually symptom-free when breastfed because of the low protein concentration in human milk, but develop vomiting and diarrhea after weaning. The patients show failure to thrive, poor appetite, growth retardation, enlarged liver and spleen, prominent osteoporosis and osteopenia,[3] delayed bone age and spontaneous protein aversion. Forced feeding of protein may lead to convulsions and coma. Mental development is normal if prolonged episode of hyperammonemia can be avoided. Some patients develop severe pulmonary and renal complications. High levels of plasma glutamine and glycine are observed.
Treatment and prognosis
Treatment of LPI consists of protein-restricted diet and supplementation with oral citrulline. Citrulline is a neutral amino acid that improves the function of the urea cycle and allows sufficient protein intake without hyperammonemia. Under proper dietary control and supplementation, the majority of the LPI patients are able to have a nearly normal life. However, severe complications including pulmonary alveolar proteinosis and renal insufficiency may develop even with proper treatment. [4]
Fertility appears to be normal in women, but mothers with LPI have an increased risk for complications during pregnancy and delivery. [5]
References
- ^ Simell O, Perheentupa J, Rapola J, Visakorpi JK, Eskelin LE (August 1975). "Lysinuric protein intolerance" (Free full text). The American journal of medicine 59 (2): 229–240. doi:10.1016/0002-9343(75)90358-7. PMID 1155480. http://toxnet.nlm.nih.gov/cgi-bin/sis/search/r?dbs+hsdb:@term+@rn+56-41-7.
- ^ Borsani G, Bassi MT, Sperandeo MP, et al. (March 1999). "SLC7A7, encoding a putative permease-related protein, is mutated in patients with lysinuric protein intolerance". Nat. Genet. 21 (3): 297–301. doi:10.1038/6815. PMID 10080183.
- ^ Online 'Mendelian Inheritance in Man' (OMIM) 222700
- ^ Tanner LM, Näntö-Salonen K, Niinikoski H, et al. (June 2007). "Nephropathy advancing to end-stage renal disease: a novel complication of lysinuric protein intolerance". J Pediatr. 150 (6): 161–164. doi:10.1016/j.jpeds.2007.01.043. PMID 17517249.
- ^ Tanner LM, Näntö-Salonen K, Niinikoski H, et al. (February 2006). "Hazards associated with pregnancies and deliveries in lysinuric protein intolerance". Metabolism 55 (2): 224–231. doi:10.1016/j.metabol.2005.08.016. PMID 16423630.
External links
Inborn error of amino acid metabolism (E70–E72, 270) K→acetyl-CoA Lysine/straight chainHypertryptophanemiaG G→pyruvate→citrateG→glutamate→
α-ketoglutarateGlutamate/glutamineG→fumarateType II tyrosinemia · Type III tyrosinemia/Hawkinsinuria · Alkaptonuria/Ochronosis · Type I tyrosinemiaTransport/
IE of RTTSolute carrier family: Cystinuria · Hartnup disease · Lysinuric protein intolerance · IminoglycinuriaOther Genetic disorder, membrane: Solute carrier disorders 1-10 SLC1A3 (Episodic ataxia 6) · SLC2A1 (De Vivo disease) · SLC2A5 (Fructose malabsorption) · SLC2A10 (Arterial tortuosity syndrome) · SLC3A1 (Cystinuria) · SLC4A1 (Hereditary spherocytosis 4/Hereditary elliptocytosis 4) · SLC4A11 (Congenital endothelial dystrophy type 2, Fuchs' dystrophy 4) · SLC5A1 (Glucose-galactose malabsorption) · SLC5A2 (Renal glycosuria) · SLC5A5 (Thyroid dyshormonogenesis type 1) · SLC6A19 (Hartnup disease) · SLC7A7 (Lysinuric protein intolerance) · SLC7A9 (Cystinuria)11-20 SLC11A1 (Crohn's disease) · SLC12A3 (Gitelman syndrome) · SLC16A1 (HHF7) · SLC16A2 (Allan–Herndon–Dudley syndrome) · SLC17A5 (Salla disease) · SLC17A8 (DFNA25)21-40 see also solute carrier family
B structural (perx, skel, cili, mito, nucl, sclr) · DNA/RNA/protein synthesis (drep, trfc, tscr, tltn) · membrane (icha, slcr, atpa, abct, othr) · transduction (iter, csrc, itra), trfkCategories:- Amino acid metabolism disorders
- Autosomal recessive disorders
- Membrane transport protein disorders
- Rare diseases

Wikimedia Foundation. 2010.