- Cystinuria
-
Cystinuria Classification and external resources 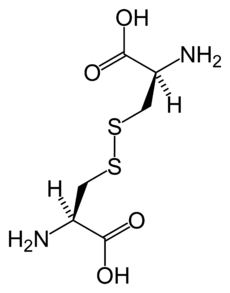
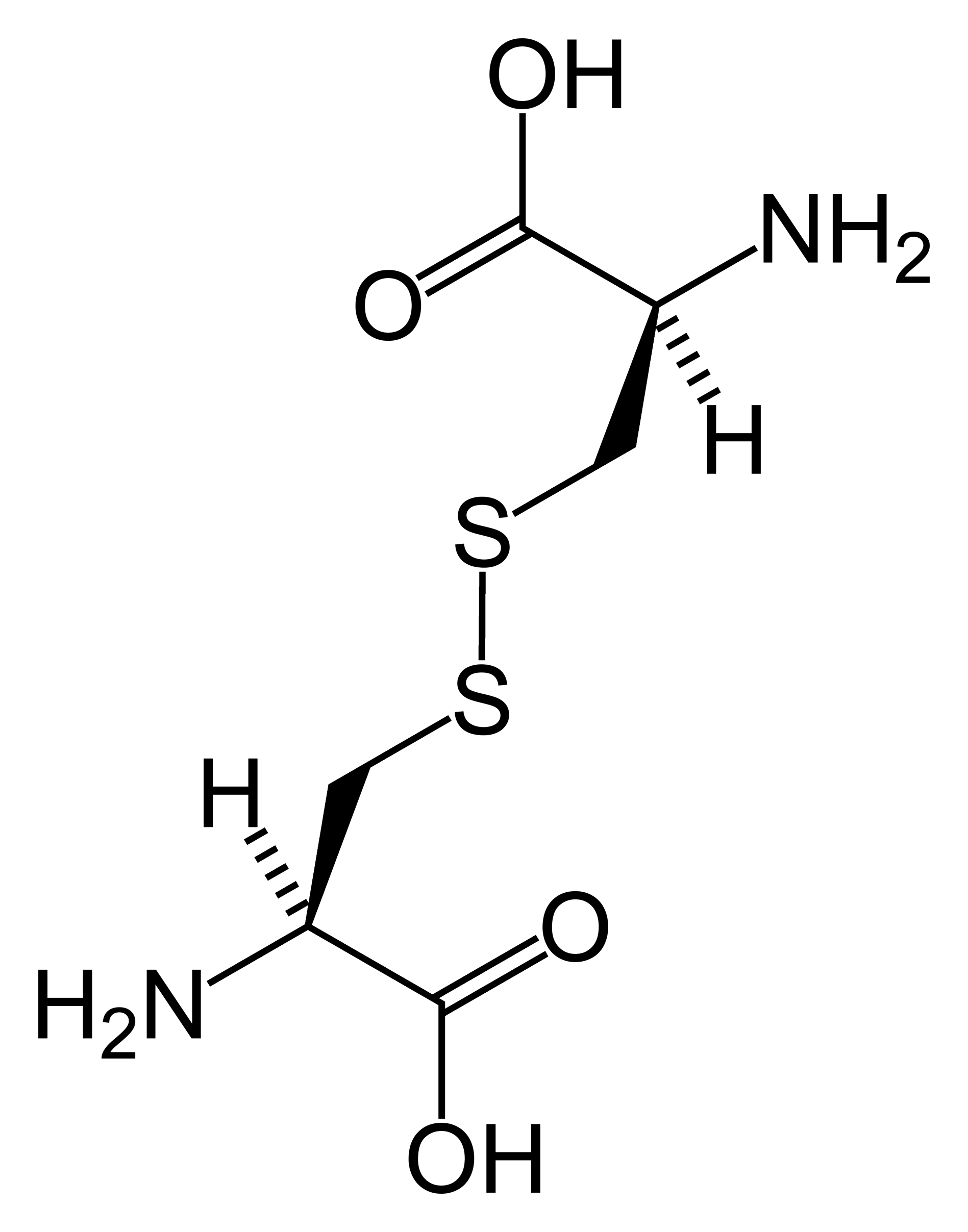
Chemical structure of cystine formed from L-cysteine (under biological conditions)ICD-10 E72.0 ICD-9 270.0 OMIM 220100 DiseasesDB 3339 MedlinePlus 000346 eMedicine med/498 MeSH D003555 Cystinuria is an inherited autosomal recessive[1] disease that is characterized by the formation of cystine stones in the kidneys, ureter, and bladder.
Contents
Signs and symptoms
Cystinuria is a cause of persistent kidney stones. It is a disease involving the defective transepithelial transport of cystine and dibasic amino acids in the kidney and intestine, and is one of many causes of kidney stones. If not treated properly, the disease could cause serious damage to the kidneys and surrounding organs, and in some rare cases death. The stones may be identified by a positive nitroprusside cyanide test. The crystals are usually hexagonal, translucent, white. Upon removal, the stones may be pink or yellow in color, but later they turn to greenish due to exposure to air.
Genetics
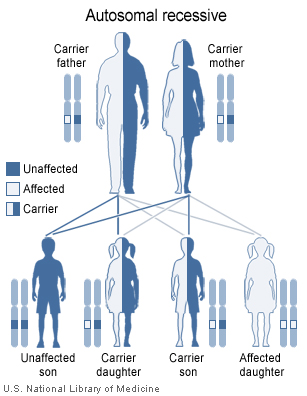
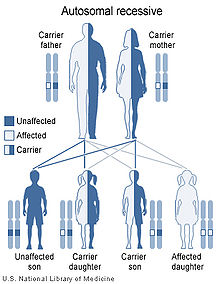 Cystinuria has an autosomal recessive pattern of inheritance.
Cystinuria has an autosomal recessive pattern of inheritance.
Cystinuria is an autosomal recessive disease,[1] which means that the defective gene responsible for the disease is located on an autosome, and two copies of the defective gene (one inherited from each parent) are required in order to be born with the disease. The parents of an individual with an autosomal recessive disease both carry one copy of the defective gene, but usually do not experience any signs or symptoms of the disease.
Cause
Cystinuria is caused by mutations in the SLC3A1 and SLC7A9 genes. These genes encode two parts of a transporter protein that is made primarily in the kidneys. These defects prevent proper reabsorption of basic, or positively charged, amino acids: lysine, ornithine, arginine.[2] Under normal circumstances, this protein allows certain amino acids, including cysteine, to be reabsorbed into the blood from the filtered fluid that will become urine. Mutations in either of these genes disrupt the ability of this transporter protein to reabsorb these amino acids, allowing them to become concentrated in the urine. As the levels of cystine in the urine increase, the crystals typical of cystinuria are able to form, resulting in kidney stones. Cystine crystals form hexagonal-shaped crystals that can be viewed upon microscopic analysis of the urine. The other amino acids that are not reabsorbed do not create crystals in urine. The disease affects 1 in 10,000 people, making it the most common genetic error of amino acid transport. Cystinuria is inherited in an autosomal recessive pattern.
Pathophysiology
Cystinuria is characterized by the inadequate reabsorption of cystine in the proximal convoluted tubules after the filtering of the amino acids by the kidney's glomeruli, thus resulting in an excessive concentration of this amino acid in the urine. Cystine may precipitate out of the urine, if the urine is neutral or acidic, and form crystals or stones in the kidneys, ureters, or bladder. It is one of several inborn errors of metabolism included in the Garrod's tetrad. The disease is attributed to deficiency in transport and metabolism of amino acids.
Clinical Features
Cystinuria is usually asymptomatic when no stone is formed. However, once a stone is formed, or if stone production is severe or frequent, the following symptoms may be present:
- Nausea/Vomiting
- Dull ache or "colicky" pain
- Chronic pain
- Hematuria
- Obstructive syndromes like hydronephrosis
- Infective syndromes like pyelonephritis
- Blood in urine (blood in the urine is not always present in some cystinurics)
Cystinurics can also experience chronic pain in one, or both, kidneys[1] due to the scars that the jagged edges of the stones can leave or damage from multiple stone removal surgeries[2]. This can leave a cystinuric in constant pain which often requires medical intervention, such as long term use of analgesics or surgical procedures, including T11, T12 or T13 nerve blocks (although, these procedures are often not successful, they can provide some relief). Aside from the chronic pain, a cystinuric will often have severe breakthrough pain from passing stones. This type of severe pain, if not properly treated, will force the cystinuric to seek help in an Emergency Department or Urgent Care facility. Since most cystinurics pass stones monthly, weekly, or daily, and need ongoing care, this usually causes medical staff to label these patients as drug seekers or hypochondriacs. Cystinurics have an increased risk for chronic kidney disease[3][4] and since kidney damage or poor function is often present in cystinurics, the use of nonsteroidal anti-inflammatory drugs (NSAIDs) or over the counter (OTC) medications should be used with caution. This leaves the patient with little choices regarding pain control and can lead to insufficient medical intervention by physicians.
Cystine stones are often not visible on most x-rays, CT's, and ultrasounds[5]. This does not mean the cystinuric doesn't have a stone. It takes a trained eye and experience to spot a cystine stone. It is not unusual for a cystinuric to pass a stone, or stones, after being released from the hospital with a CT or x-ray result of no stones in the kidneys.
Investigations
1. Blood: Routine hemogram along with blood sugar, urea, and creatinine.
2. Urine: For cystine crystals, and casts.
3. USG/CT scan to reveal if a stone is present.
4. Genetic analysis to determine which mutation associated with the disease may be present. Currently genotyping is not available in the United States but might be available in Spain and Italy.
Regular x-rays often fail to show the cystine stones. Cystine stones seem to be 'invisible' with this method unless a dye is added to the blood stream (IVP). ([Pyelogram])
Treatment
Initial treatment is with adequate hydration, alkalinization of the urine with citrate supplementation, and dietary modification to reduce salt and protein intake. If this fails then patients are usually started on chelation therapy with an agent such as penicillamine.[3][4] Once renal stones have formed, however, the first-line treatment is surgery. Both endoscopic surgery and conventional open-abdominal surgery have proven to be effective treatment modalities for patients with more advanced disease.
Jeffrey Rimer of the University of Houston suggested in October 2010 that treatment with L-cystine dimethylester could prevent the formation of these kidney stones.[5]
Videos of surgery are available on various websites that show stone removal by Percutaneous Nephrolithotomy.
Occurrence in animals
This disease is known to occur in at least three mammalian species. These are: humans, domestic dogs canines, and a wild canid, the Maned Wolf of South America. Cystine uroliths have been demonstrated, most usually in male dogs, from approximately 70 breeds including the Australian cattle dog, Australian shepherd, Basenji, Basset, Bullmastiff, Chihuahua, Scottish deerhound, Scottish terrier, Staffordshire terrier and Welsh corgi; and in both male and female Newfoundland dogs [6]
See also
- Cystine
- Cysteine
- Thiola
- International Cystinuria Foundation
- Hartnup disease
- Cystinosis
- Homocystinuria
References
- ^ a b Fjellstedt E, Harnevik L, Jeppsson JO, Tiselius HG, Söderkvist P, Denneberg T (Dec 2003). "Urinary excretion of total cysteine and the dibasic amino acids arginine, lysine and ornithine in relation to genetic findings in patients with cystinuria treated with sulfhydryl compounds". Urological research 31 (6): 417–425. doi:10.1007/s00240-003-0366-6. PMID 14586528.
- ^ Ahmed K, Dasgupta P, Khan MS (2006). "Cystine calculi: challenging group of stones". Postgraduate medical journal 82 (974): 799–801. doi:10.1136/pgmj.2005.044156. PMC 2653923. PMID 17148700. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2653923.
- ^ Ahmed K, Khan MS, Thomas K, Challacombe B, Bultitude M, Glass J, Tiptaft R, Dasgupta P (Mar 2008). "Management of cystinuric patients: an observational, retrospective, single-centre analysis". Urol Int. 80 (2): 141–144. doi:10.1159/000112603. PMID 18362482.
- ^ Joly D, Rieu P, Méjean A, Gagnadoux MF, Daudon M, Jungers P (November 1999). "Treatment of cystinuria". Pediatr. Nephrol. 13 (9): 945–50. doi:10.1007/s004670050736. PMID 10603157. http://link.springer.de/link/service/journals/00467/bibs/9013009/90130945.htm.
- ^ Science 15 October 2010, Vol. 330 no. 6002 pp. 337-341 DOI: 10.1126/science.1191968
- ^ D Bannasch and PS Henthorn (2009). "Changing Paradigms in Diagnosis of Inherited Defects Associated with Uroliths". Veterinary Clinics of North America Small Animal Practice 39 (1): 111–125. doi:10.1016/j.cvsm.2008.09.006. PMC 2628803. PMID 19038654. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2628803.
External links
Inborn error of amino acid metabolism (E70–E72, 270) K→acetyl-CoA Lysine/straight chainHypertryptophanemiaG G→pyruvate→citrateG→glutamate→
α-ketoglutarateGlutamate/glutamineG→fumarateType II tyrosinemia · Type III tyrosinemia/Hawkinsinuria · Alkaptonuria/Ochronosis · Type I tyrosinemiaTransport/
IE of RTTOther Genetic disorder, membrane: Solute carrier disorders 1-10 SLC1A3 (Episodic ataxia 6) · SLC2A1 (De Vivo disease) · SLC2A5 (Fructose malabsorption) · SLC2A10 (Arterial tortuosity syndrome) · SLC3A1 (Cystinuria) · SLC4A1 (Hereditary spherocytosis 4/Hereditary elliptocytosis 4) · SLC4A11 (Congenital endothelial dystrophy type 2, Fuchs' dystrophy 4) · SLC5A1 (Glucose-galactose malabsorption) · SLC5A2 (Renal glycosuria) · SLC5A5 (Thyroid dyshormonogenesis type 1) · SLC6A19 (Hartnup disease) · SLC7A7 (Lysinuric protein intolerance) · SLC7A9 (Cystinuria)11-20 SLC11A1 (Crohn's disease) · SLC12A3 (Gitelman syndrome) · SLC16A1 (HHF7) · SLC16A2 (Allan–Herndon–Dudley syndrome) · SLC17A5 (Salla disease) · SLC17A8 (DFNA25)21-40 see also solute carrier family
B structural (perx, skel, cili, mito, nucl, sclr) · DNA/RNA/protein synthesis (drep, trfc, tscr, tltn) · membrane (icha, slcr, atpa, abct, othr) · transduction (iter, csrc, itra), trfkCategories:- Amino acid metabolism disorders
- Autosomal recessive disorders
- Membrane transport protein disorders
Wikimedia Foundation. 2010.