- Isobutyryl-coenzyme A dehydrogenase deficiency
Infobox_Disease
Name = PAGENAME
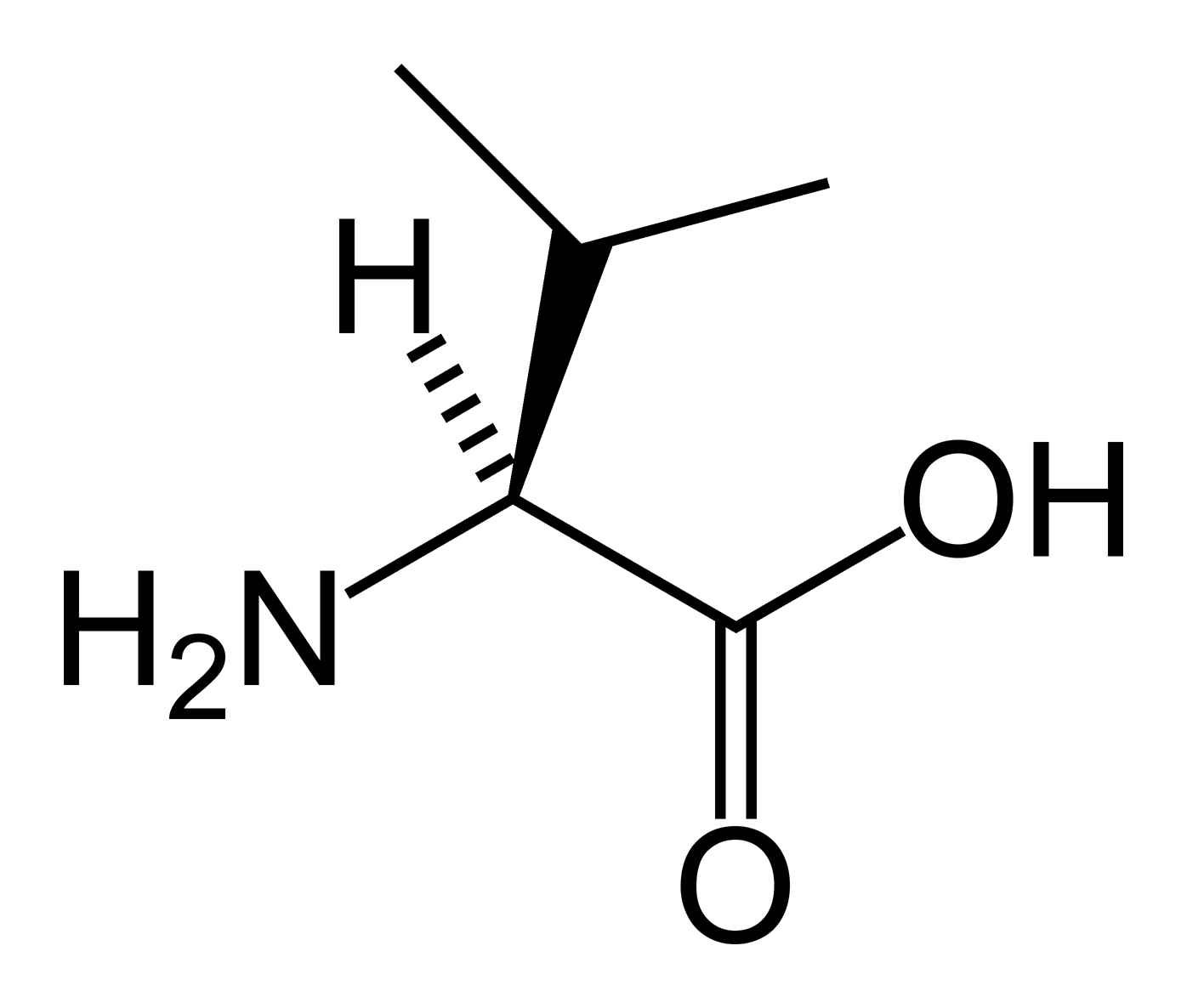
Caption =Valine
DiseasesDB = 34225
ICD10 =
ICD9 =
ICDO =
OMIM = 604773
MedlinePlus =
eMedicineSubj =
eMedicineTopic =
MeshID =Isobutyryl-coenzyme A dehydrogenase deficiency, commonly known as IBD deficiency, is a rare
metabolic disorder in which the body is unable to process certainamino acid s properly.People with this disorder have inadequate levels of an enzyme that helps break down the amino acid
valine , resulting in a build up of valine in the urine, a symptom calledvalinuria .Diagnosis
Babies with this disorder are usually healthy at birth. The signs and symptoms may not appear until later in infancy or childhood and can include poor feeding and growth (failure to thrive), a weakened and enlarged heart (
dilated cardiomyopathy ), seizures, and low numbers of red blood cells (anemia ). Another feature of this disorder may be very low blood levels ofcarnitine (a natural substance that helps convert certain foods into energy).Isobutyryl-CoA dehydrogenase deficiency may be worsened by long periods without food (fasting) or infections that increase the body's demand for energy. Some individuals with gene mutations that can cause isobutyryl-CoA dehydrogenase deficiency may never experience any signs and symptoms of the disorder.
Genetics
Defects in the "
ACAD8 " gene cause isobutyryl-coenzyme A dehydrogenase deficiency. The "ACAD8" gene provides instructions for making an enzyme that plays an essential role in breaking down proteins from the diet. Specifically, the enzyme is responsible for processing valine, an amino acid that is part of many proteins. If a mutation in the ACAD8 gene reduces or eliminates the activity of this enzyme, the body is unable to break down valine properly. As a result, poor growth and reduced energy production may occur.This disorder is inherited in an
autosomal recessive pattern, which means the defective gene is located on anautosome , and two copies of the gene - one from each parent - are needed to be born with the disorder. The parents of an individual with an autosomal recessive disorder are carriers of one copy of the defective gene, but do not show signs and symptoms of the disorder.ee also
*
Isobutyryl-coenzyme A External links
*

Wikimedia Foundation. 2010.