- Methylmalonic acidemia
-
Methylmalonic acidemia Classification and external resources 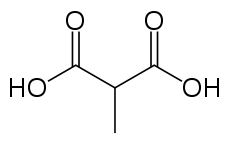
Methylmalonic acidICD-10 E71.1 ICD-9 270.3 OMIM 251000 251100 251110 277380 277400 277410 606169 DiseasesDB 29509 29510 MedlinePlus 001162 eMedicine neuro/576 Methylmalonic acidemia (MMA), also called methylmalonic aciduria, is an autosomal recessive[1] metabolic disorder[2]. It is a classical type of organic acidemia.[3]
Methylmalonic acidemia stems from several genotypes,[4] all forms of the disorder usually diagnosed in the early neonatal period, presenting progressive encephalopathy, and secondary hyperammonemia. The disorder can result in death if undiagnosed or left untreated.
Contents
Causes
Genetic
 Methylmalonic acidemia has an autosomal recessive pattern of inheritance.
Methylmalonic acidemia has an autosomal recessive pattern of inheritance.
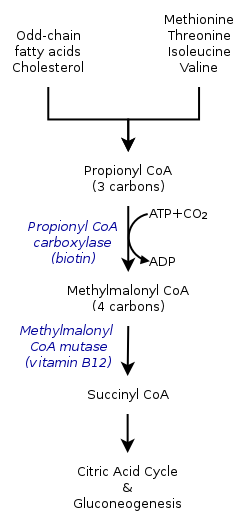
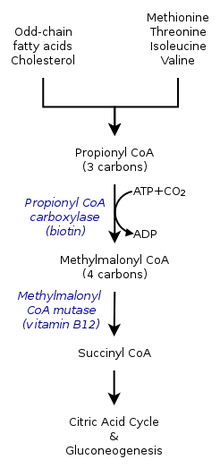 Methylmalonic acidemia is caused by a defect in the vitamin B12-dependent enzyme methylmalonyl CoA mutase.
Methylmalonic acidemia is caused by a defect in the vitamin B12-dependent enzyme methylmalonyl CoA mutase.The inherited forms of methylmalonic acidemia cause defects in the metabolic pathway where methylmalonyl-coenzyme A (CoA) is converted into succinyl-CoA by the enzyme methylmalonyl-CoA mutase.[5]
Vitamin B12 is also needed for the conversion of methylmalonyl-CoA to Succinyl-CoA. Mutations leading to defects in vitamin B12 metabolism or in its transport frequently result in the development of methylmalonic acidemia.
This disorder has an autosomal recessive inheritance pattern, which means the defective gene is located on an autosome, and two copies of the gene - one from each parent - must be inherited to be affected by the disorder. The parents of a child with an autosomal recessive disorder are carriers of one copy of the defective gene, but are usually not affected by the disorder.
Nutritional
A severe nutritional deficiency of vitamin B12 can also result in methylmalonic acidemia.[6] Methylmalonyl CoA requires vitamin B12 to form succinyl-CoA. When the amount of B12 is insufficient for the conversion of cofactor methylmalonyl-CoA into succinyl-CoA, the buildup of unused methylmalonyl-CoA eventually leads to methylmalonic acidemia. This diagnosis is often used as an indicator of vitamin B12 deficiency in serum.[7]
Pathogenesis
Methylmalonic acidemia has varying diagnoses, treatment requirements and prognoses, which are determined by the specific genetic mutation causing the inherited form of the disorder.[4] The following are the known genotypes responsible for methylmalonic acidemia:
OMIM Name Gene 251100 cblA type MMAA 251110 cblB type MMAB 277400 cblC type MMACHC 277410 cblD type MMADHC[8] 277380 cblF type LMBRD1[9] 251000 mut type MUT See also
References
- ^ Radmanesh, A; Zaman, T; Ghanaati, H; Molaei, S; Robertson, Rl; Zamani, Aa (July 2008). "Methylmalonic acidemia: brain imaging findings in 52 children and a review of the literature". Pediatric radiology 38 (10): 1054. doi:10.1007/s00247-008-0940-8. PMID 18636250.
- ^ http://www.genome.gov/19016901
- ^ Dionisi-Vici C, Deodato F, Raschinger W, Rhead W, Wilcken B (2006). "Classical organic acidurias, propionic aciduria, methylmalonic aciduria, and isovaleric aciduria: long-term outcome and effects of expanded newborn screening using tandem mass spectrometry". J Inherit Metab Dis. 29 (2-3): 383–389. doi:10.1007/s10545-006-0278-z. PMID 16763906.
- ^ a b Matsui, Sm; Mahoney, Mj; Rosenberg, Le (April 1983). "The natural history of the inherited methylmalonic acidemias" (Free full text). The New England journal of medicine 308 (15): 857–61. doi:10.1056/NEJM198304143081501. ISSN 0028-4793. PMID 6132336. http://toxnet.nlm.nih.gov/cgi-bin/sis/search/r?dbs+hsdb:@term+@rn+68-19-9.
- ^ Sakomoto O, Ohura T, Matsubara Y, Takayanagi M, Tsuchiya S (2007). "Mutation and haplotype analyses of the MUT gene in Japanese patients with methylmalonic acidemia". J Hum Genet. 52 (1): 48–55. doi:10.1007/s10038-006-0077-2. PMID 17075691.
- ^ Higginbottom MC, Sweetman L, Nyhan WL (1978). "A syndrome of methylmalonic aciduria, homocystinuria, megaloblastic anemia and neurological abnormalities in a vitamin B12-deficient breast-fed infant of a strict vegetarian". N Engl J Med. 299 (7): 317–323. doi:10.1056/NEJM197808172990701. PMID 683264.
- ^ http://www.biology.arizona.edu/biochemistry/problem_sets/b12/04t.html
Vitamin B12 deficiency - The methylmalonic aciduria connection - ^ Gene identification for the cblD defect of vitamin B12 metabolism. Coelho D, Suormala T, Stucki M, Lerner-Ellis JP, Rosenblatt DS, Newbold RF, Baumgartner MR, Fowler B. N Engl J Med. 2008 Apr 3;358(14):1454-64. PMID: 18385497
- ^ Identification of a putative lysosomal cobalamin exporter altered in the cblF defect of vitamin B12 metabolism. Rutsch F, Gailus S, Miousse IR, Suormala T, Sagné C, Toliat MR, Nürnberg G, Wittkampf T, Buers I, Sharifi A, Stucki M, Becker C, Baumgartner M, Robenek H, Marquardt T, Höhne W, Gasnier B, Rosenblatt DS, Fowler B, Nürnberg P. Nat Genet. 2009 Feb;41(2):234-9. Epub 2009 Jan 11. PMID: 19136951
External links
- Organic Acidemia Association
- Methylmalonic acidemia at NLM Genetics Home Reference
- Washington Health Center
- GeneReviews article on Methylmalonic Acidemia
- GeneReviews article on Disorders of Intracellular Cobalamin Metabolism
- [1]
Inborn error of amino acid metabolism (E70–E72, 270) K→acetyl-CoA Lysine/straight chainHypertryptophanemiaG G→pyruvate→citrateG→glutamate→
α-ketoglutarateGlutamate/glutamineG→fumarateType II tyrosinemia · Type III tyrosinemia/Hawkinsinuria · Alkaptonuria/Ochronosis · Type I tyrosinemiaTransport/
IE of RTTOther Metabolic disorders of vitamins, coenzymes, and cofactors B7 Biotin/MCD Other B B5 (Pantothenate kinase-associated neurodegeneration) · B12 (Methylmalonic acidemia)Other vitamin Nonvitamin cofactor M: NUT
cof, enz, met
noco, nuvi, sysi/epon, met
drug(A8/11/12)
Categories:- Autosomal recessive disorders
- Amino acid metabolism disorders
- Rare diseases
- Vitamin, coenzyme, and cofactor metabolism disorders
- Genetic disorder stubs


![[1]](http://www.newbornscreening.info/Parents/organicaciddisorders/images/MMA.gif&imgrefurl=http://www.newbornscreening.info/Parents/organic){kind=link}
Wikimedia Foundation. 2010.