- Methylmalonyl-CoA mutase
-
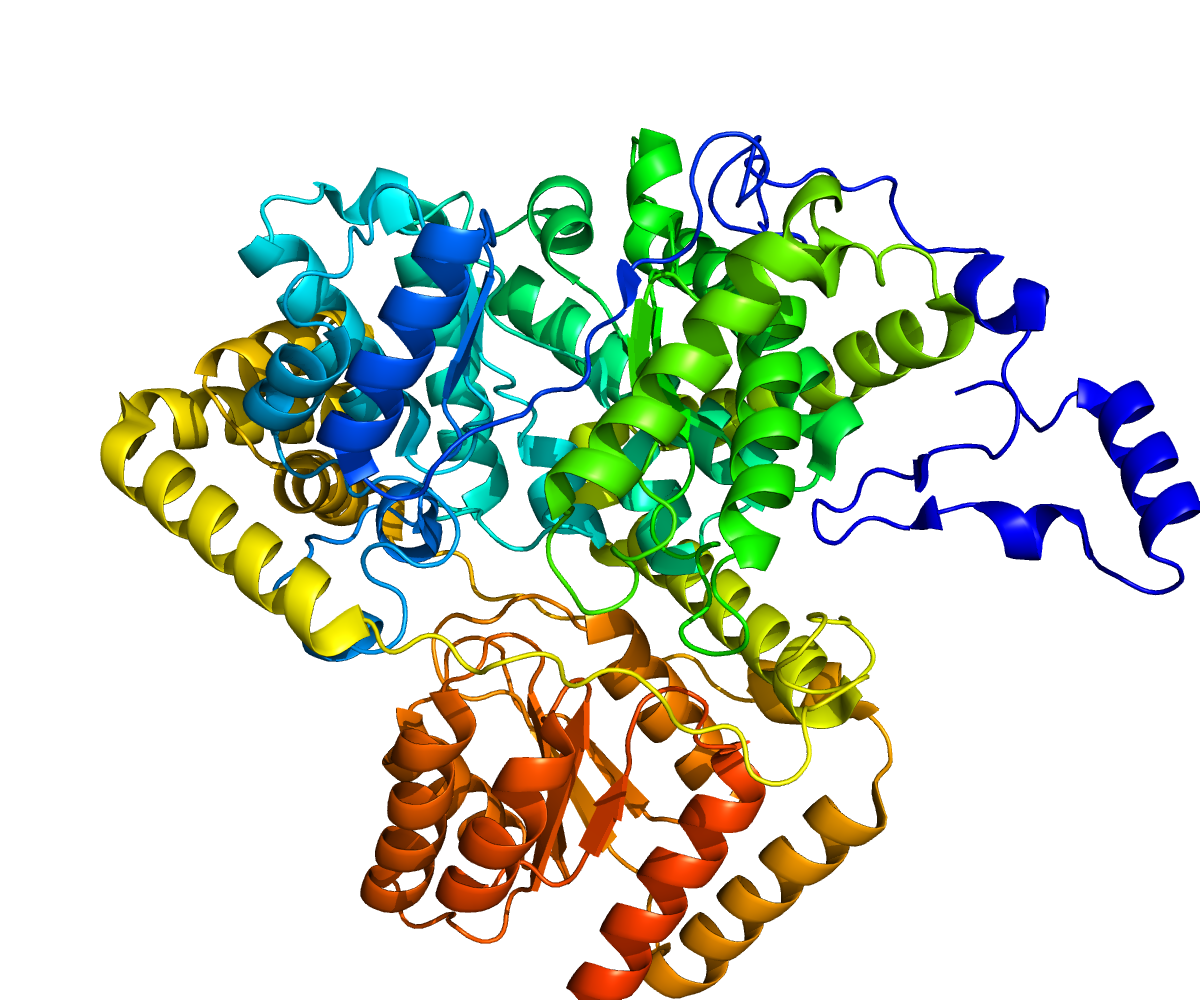
Methylmalonyl CoA mutase 
Rendering based on PDB 2XIJ.Available structures PDB 2XIJ, 2XIQ, 3BIC Identifiers Symbols MUT; MCM External IDs OMIM: 609058 MGI: 97239 HomoloGene: 20097 GeneCards: MUT Gene EC number 5.4.99.2 Gene Ontology Molecular function • methylmalonyl-CoA mutase activity
• cobalamin binding
• metal ion bindingCellular component • mitochondrion
• mitochondrial matrixBiological process • fatty acid beta-oxidation
• post-embryonic development
• cellular lipid metabolic processSources: Amigo / QuickGO RNA expression pattern 
More reference expression data Orthologs Species Human Mouse Entrez 4594 17850 Ensembl ENSG00000146085 ENSMUSG00000023921 UniProt P22033 Q3UFU2 RefSeq (mRNA) NM_000255.3 NM_008650.3 RefSeq (protein) NP_000246.2 NP_032676.2 Location (UCSC) Chr 6:
49.4 – 49.43 MbChr 17:
41.07 – 41.1 MbPubMed search [1] [2] methylmalonyl-CoA mutase Identifiers EC number 5.4.99.2 CAS number 9023-90-9 Databases IntEnz IntEnz view BRENDA BRENDA entry ExPASy NiceZyme view KEGG KEGG entry MetaCyc metabolic pathway PRIAM profile PDB structures RCSB PDB PDBe PDBsum Gene Ontology AmiGO / EGO Search PMC articles PubMed articles Methylmalonyl Coenzyme A mutase, also known as MCM is an enzyme that catalyzes the isomerization of methylmalonyl-CoA to succinyl-CoA and it is involved in key metabolic pathways. It requires a vitamin B12-derived prosthetic group, adenosylcobalamin, to function.
Contents
Function
The substrate of methylmalonyl-CoA mutase, methylmalonyl-CoA, is primarily derived from propionyl-CoA, a substance formed from the catabolism and digestion of isoleucine, valine, threonine, methionine, thymine, cholesterol, or odd-chain fatty acids.
The product of the enzyme, succinyl-CoA, is a key molecule of the TCA cycle.
MCM resides in the mitochondria, where a number of substances, including the branched-chain amino acids isoleucine and valine, as well as methionine, threonine, thymine and odd-chain fatty acids, are metabolized via methylmalonate semialdehyde (MMlSA) or propionyl-CoA (Pr-CoA) to a common compound - methylmalonyl-CoA (MMl-CoA).
Human Genetics
The gene encoding for this enzyme in humans is known as MUT[1].
Pathology
A deficiency of this enzyme is responsible for an inherited disorder of metabolism, Methylmalonyl-CoA mutase deficiency, which is one of the causes of methylmalonic acidemia.
Mechanism
MUT reaction mechanism begins with homolytic cleavage of AdoB12's C-Co(III) bond, the C and Co atoms each acquire one of the electrons that formed the cleaved electron pair bond. The Co ion, therefore, fluctuates between its Co(III) and Co(II) oxidation states [the two states are spectroscopically distinguishable: Co(III) is red and diamagnetic (no unpaired electrons), whereas Co(II) is yellow and paramagnetic (unpaired electrons)]. Hence, the role of coenzyme B-12 in the catalytic process is that of a reversible free radical generator. The C-Co(III) bond is well suited to this function because it is inherently weak (dissociation energy = 109 kJ/mol) and appears to be further weakened through steric interactions with the enzyme. A homolytic cleavage reaction is unusual in biology; most other biological bond cleavage reactions occur via heterolytic cleavage (in which the electron pair forming the cleaved bond is fully acquired by one of the separating atoms).

See also
- MMAA
- MMAB
- MMACHC
- MMADHC
References
Further reading
- Ledley FD, Rosenblatt DS (1997). "Mutations in mut methylmalonic acidemia: clinical and enzymatic correlations.". Hum. Mutat. 9 (1): 1–6. doi:10.1002/(SICI)1098-1004(1997)9:1<1::AID-HUMU1>3.0.CO;2-E. PMID 8990001.
- Ludwig ML, Matthews RG (1997). "Structure-based perspectives on B12-dependent enzymes.". Annu. Rev. Biochem. 66: 269–313. doi:10.1146/annurev.biochem.66.1.269. PMID 9242908.
- Lubrano R, Elli M, Rossi M, et al. (2007). "Renal transplant in methylmalonic acidemia: could it be the best option? Report on a case at 10 years and review of the literature.". Pediatr. Nephrol. 22 (8): 1209–14. doi:10.1007/s00467-007-0460-z. PMID 17401587.
- Frenkel EP, Kitchens RL (1978). "Intracellular localization of hepatic propionyl-CoA carboxylase and methylmalonyl-CoA mutase in humans and normal and vitamin B12 deficient rats.". Br. J. Haematol. 31 (4): 501–13. doi:10.1111/j.1365-2141.1975.tb00885.x. PMID 24458.
- Crane AM, Jansen R, Andrews ER, Ledley FD (1992). "Cloning and expression of a mutant methylmalonyl coenzyme A mutase with altered cobalamin affinity that causes mut- methylmalonic aciduria.". J. Clin. Invest. 89 (2): 385–91. doi:10.1172/JCI115597. PMC 442864. PMID 1346616. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=442864.
- Crane AM, Martin LS, Valle D, Ledley FD (1992). "Phenotype of disease in three patients with identical mutations in methylmalonyl CoA mutase.". Hum. Genet. 89 (3): 259–64. doi:10.1007/BF00220536. PMID 1351030.
- Raff ML, Crane AM, Jansen R, et al. (1991). "Genetic characterization of a MUT locus mutation discriminating heterogeneity in mut0 and mut- methylmalonic aciduria by interallelic complementation.". J. Clin. Invest. 87 (1): 203–7. doi:10.1172/JCI114972. PMC 295026. PMID 1670635. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=295026.
- Jansen R, Ledley FD (1990). "Heterozygous mutations at the mut locus in fibroblasts with mut0 methylmalonic acidemia identified by polymerase-chain-reaction cDNA cloning.". Am. J. Hum. Genet. 47 (5): 808–14. PMC 1683687. PMID 1977311. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1683687.
- Nham SU, Wilkemeyer MF, Ledley FD (1991). "Structure of the human methylmalonyl-CoA mutase (MUT) locus.". Genomics 8 (4): 710–6. doi:10.1016/0888-7543(90)90259-W. PMID 1980486.
- Ledley FD, Lumetta M, Nguyen PN, et al. (1988). "Molecular cloning of L-methylmalonyl-CoA mutase: gene transfer and analysis of mut cell lines.". Proc. Natl. Acad. Sci. U.S.A. 85 (10): 3518–21. doi:10.1073/pnas.85.10.3518. PMC 280243. PMID 2453061. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=280243.
- Jansen R, Kalousek F, Fenton WA, et al. (1989). "Cloning of full-length methylmalonyl-CoA mutase from a cDNA library using the polymerase chain reaction.". Genomics 4 (2): 198–205. doi:10.1016/0888-7543(89)90300-5. PMID 2567699.
- Fenton WA, Hack AM, Kraus JP, Rosenberg LE (1987). "Immunochemical studies of fibroblasts from patients with methylmalonyl-CoA mutase apoenzyme deficiency: detection of a mutation interfering with mitochondrial import.". Proc. Natl. Acad. Sci. U.S.A. 84 (5): 1421–4. doi:10.1073/pnas.84.5.1421. PMC 304442. PMID 2881300. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=304442.
- Zoghbi HY, O'Brien WE, Ledley FD (1989). "Linkage relationships of the human methylmalonyl CoA mutase to the HLA and D6S4 loci on chromosome 6.". Genomics 3 (4): 396–8. doi:10.1016/0888-7543(88)90135-8. PMID 2907507.
- Kolhouse JF, Utley C, Allen RH (1980). "Isolation and characterization of methylmalonyl-CoA mutase from human placenta.". J. Biol. Chem. 255 (7): 2708–12. PMID 6102092.
- Fenton WA, Hack AM, Willard HF, et al. (1982). "Purification and properties of methylmalonyl coenzyme A mutase from human liver.". Arch. Biochem. Biophys. 214 (2): 815–23. doi:10.1016/0003-9861(82)90088-1. PMID 6124211.
- Qureshi AA, Crane AM, Matiaszuk NV, et al. (1994). "Cloning and expression of mutations demonstrating intragenic complementation in mut0 methylmalonic aciduria.". J. Clin. Invest. 93 (4): 1812–9. doi:10.1172/JCI117166. PMC 294249. PMID 7909321. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=294249.
- Crane AM, Ledley FD (1994). "Clustering of mutations in methylmalonyl CoA mutase associated with mut- methylmalonic acidemia.". Am. J. Hum. Genet. 55 (1): 42–50. PMC 1918235. PMID 7912889. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1918235.
- Janata J, Kogekar N, Fenton WA (1998). "Expression and kinetic characterization of methylmalonyl-CoA mutase from patients with the mut- phenotype: evidence for naturally occurring interallelic complementation.". Hum. Mol. Genet. 6 (9): 1457–64. doi:10.1093/hmg/6.9.1457. PMID 9285782.
External links
Isomerase: mutases (EC 5.4) 5.4.2 Phosphomutases 5.4.99 Other groups Methylmalonyl-CoA mutase · Lanosterol synthaseB enzm: 1.1/2/3/4/5/6/7/8/10/11/13/14/15-18, 2.1/2/3/4/5/6/7/8, 2.7.10, 2.7.11-12, 3.1/2/3/4/5/6/7, 3.1.3.48, 3.4.21/22/23/24, 4.1/2/3/4/5/6, 5.1/2/3/4/99, 6.1-3/4/5-6 Metabolism: amino acid metabolism · synthesis and catabolism enzymes (essential in CAPS) K→acetyl-CoA (see below)G G→pyruvate
→citrateG→glutamate→
α-ketoglutarate→alpha-ketoglutarate→TCAOther→succinyl-CoA→TCAG→fumarateasparagine→aspartate→Metabolism: Citric acid cycle enzymes Cycle Anaplerotic to acetyl-CoAPyruvate dehydrogenase complex (E1, E2, E3)
(regulated by Pyruvate dehydrogenase kinase and Pyruvate dehydrogenase phosphatase)to succinyl-CoAMethylmalonyl-CoA mutaseto oxaloacetateMitochondrial
electron transport chain/
oxidative phosphorylationPrimaryComplex I/NADH dehydrogenase · Complex II/Succinate dehydrogenase · Coenzyme Q · Complex III/Coenzyme Q - cytochrome c reductase · Cytochrome c · Complex IV/Cytochrome c oxidase
Coenzyme Q10 synthesis: COQ2 · COQ3 · COQ4 · COQ5 · COQ6 · COQ7 · COQ9 · COQ10A · COQ10B · PDSS1 · PDSS2OtherThis isomerase article is a stub. You can help Wikipedia by expanding it.