- Pantothenate kinase-associated neurodegeneration
-
Pantothenate kinase-associated neurodegeneration Classification and external resources 
PantetheineICD-10 G23.0 ICD-9 333.0 OMIM 234200 DiseasesDB 29462 MedlinePlus 001225 eMedicine neuro/151 MeSH D006211 GeneReviews Pantothenate Kinase-Associated Neurodegeneration Pantothenate kinase-associated neurodegeneration (PKAN), also known as neurodegeneration with brain iron accumulation 1 (NBIA1) and formerly called Hallervorden-Spatz syndrome (use of this eponym is discouraged due to Drs. Julius Hallervorden and Hugo Spatz's affiliation with the Nazi regime and the ethically unacceptable manner in which they obtained some autopsy specimens[1][2]), is a degenerative disease of the brain that can lead to parkinsonism, dystonia, dementia, and ultimately death. Neurodegeneration in PKAN is accompanied by an excess of iron that progressively builds up in the brain.
Contents
Signs and symptoms
Symptoms typically begin in childhood and are progressive, often resulting in death by early adulthood. Symptoms of PKAN begin before middle childhood, and most often are noticed before ten years of age. Symptoms include:
- dystonia (repetitive uncontrollable muscle contractions that may cause jerking or twisting of certain muscle groups)
- dysphagia & dysarthria due to muscle groups involved in speech being involved
- rigidity/stiffness of limbs
- tremor
- writhing movements
- dementia
- spasticity
- weakness
- seizures
- retinitis pigmentosa, another degenerative disease that affects the individual’s retina, often causing alteration of retinal color and progressive deterioration of the retina at first causing night blindness and later resulting in a complete loss of vision
25% of individuals experience an uncharacteristic form of PKAN that develops post-10 years of age and follows a slower, more gradual pace of deterioration than those pre-10 years of age. These individuals face significant speech deficits as well as psychiatric and behavioral disturbances.
Being a progressive, degenerative nerve illness, PKAN leads to early immobility and often death by early adulthood.
Genetics
Genetically speaking, PKAN is an autosomal recessive disorder. The parents of an afflicted child must both be heterozygous carriers for the disease and therefore must carry one mutant allele. As it is an autosomal disorder, those heterozygous for the disorder may not display any atypical characteristics that are considered suggestive of the disorder, however there have been reported cases of compound heterozygosity in which heterozygous individuals do develop the classic form of the disease.[3][4]
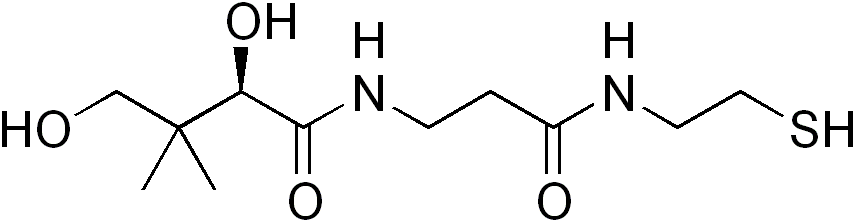
The disorder is caused by a mutant PANK2 gene located at the chromosomal locus: 20p13-p12.3. PANK2 is responsible in coding for the protein Pantothenate kinase 2, which in turn is responsible for stifling the accumulation of N-pantothenoyl-cysteine and pantetheine. It is believed that when this accumulation is not suppressed, the result is direct cell toxicity or cell toxicity as a result of free radical damage due to the lack of suppression.
PANK2 encodes a 1.85Kb transcript which is derived from seven exons covering a total distance of approximately 3.5Mb of genomic DNA. The PANK2 gene also encodes a 50.5-kDaprotein that is a functional pantothenate kinase, an essential regulatory enzyme in coenzyme A (CoA) biosynthesis, and catalyzing the phosphorylation of pantothenate (vitamin B5), N-pantothenoyl-cysteine, and pantetheine (OMIM).
Mutant PANK2 gene coded proteins are often caused by null or missense mutations most notably a 7bp deletion in the PANK2 gene coding sequence.
This disorder has been reported in specific communities based on intra-community marriages where both parents of the child are carrying the same mutation. One of the communities reported is Agrawal (Agarwal) Community mainly based in Northern Part of India. [5]
Diagnosis
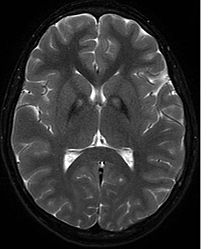 MRI image shows iron deposits in the basal ganglia, the so-called eye-of-the-tiger sign (T2w GRASE sequence).
MRI image shows iron deposits in the basal ganglia, the so-called eye-of-the-tiger sign (T2w GRASE sequence).
A neurological examination would show evidence of muscle rigidity; weakness; and abnormal postures, movements, and tremors. If other family members are also affected, this may help determine the diagnosis. Genetic tests can confirm an abnormal gene causing the disease. However, this test is not yet widely available. Other movement disorders and diseases must be ruled out. Individuals exhibiting any of the above listed symptoms are often tested using MRI (Magnetic Resonance Imaging) for a number of neuro-related disorders. As PKAN is a disease prominently evident in the brain, MRIs are very useful in making a sound diagnosis. An MRI usually shows iron deposits in the basal ganglia. Development of diagnostic criteria continues in the hope of further separating PKAN from other forms of neurodegenerative diseases featuring NBIA.
Neuropathology
Microscopic features of PKAN include:
- Iron granules
- Spheroid bodies
- Lewy bodies within neurons
Treatment
As of yet there are no major breakthroughs in the treatment of PKAN, with most pharmacologic treatments focusing on the easing or temporary relieving of PKAN’s symptoms. Iron chelating agents have been used somewhat successfully in retarding the disorder, however they have not been anywhere near what one would consider a significant success
Many believe taking certain vitamins may be beneficial, including pantothenate, Coenzyme Q, and other anti-oxidants.[citation needed]
Current research focuses on the future use of high dose pantothenate, the PANK2 enzyme substrate, in possibly alleviating symptoms as well as the further development of iron chelating agents that may be better aimed at reaching the central nervous system and working to better remove excess iron from the individual’s system.
Complications may result from the medication used to treat symptoms. Immobility from the disease can also lead to skin breakdown, respiratory infections, and blood clots, among others.
History & epidemiology
PKAN was first described by Hallervorden and Spatz (1922). Their discovery was brought about by a diagnosis of a family of 12 in which five sisters exhibited progressively increasing dementia and dysarthria. Autopsies revealed brown discolorations in different areas of the brain (particularly of interest were the globus pallidus and substantia nigra regions). Further investigation and description was brought about by Meyer (1958) who diagnosed 30 separate cases of PKAN. Meyer(1958) was followed by Elejalde et al. (1978) who described 5 affected family members and hypothesized that the disorder originated in central Europe, backing up his hypothesis with clinical and genetic analysis. Further investigation and insights were provided by Malmstrom-Groth and Kristensson (1982)[6] and Jankovic et al. (1985).[7]
Diagnosis of PKAN hit a milestone with the availability of MRIs, as well as the in-depth descriptions of those MRIs provided by Littrup and Gebarski (1985),[8] Tanfani et al. (1987),[9] Sethi et al. (1988),[10]Angelini et al. (1992),[11] Casteels et al. (1994),[12] and Malandrini et al. (1995).[13] The gene was localized to chromosome 20p by Taylor et al. (1996) [14] who suggested that this disorder should be referred to as neurodegeneration with brain iron accumulation (NBIA1) to avoid the objectionable eponym of Hallervorden-Spatz. The disease was named 'pantothenate kinase-associated neurodegeneration' or PKAN by Zhou et al. (2001)[3] who suggested the name to avoid misinterpretation and to better reflect the true nature of the disorder. Most recently Pellecchia et al. (2005) published a report of 16 patients afflicted with PKAN, confirmed by genetic analysis.[15]
Survival rates for those diagnosed with typical PKAN is 11.18 years with a standard deviation of 7.8 years. Prevalence data regarding this disorder remains incomplete, however it is estimated that anywhere between 1 in 1,000,000 to 3 in 1,000,000 individuals will be afflicted with this disorder (based upon observed cases in a population), but once again this is only an estimate as the disease is so rare it is difficult to statistically and accurately ascertain.
References
- ^ doctor/535 at Who Named It?
- ^ doctor/1063 at Who Named It?
- ^ a b Zhou B, Westaway SK, Levinson B, Johnson MA, Gitschier J, Hayflick SJ (2001). "A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome". Nat. Genet. 28 (4): 345–9. doi:10.1038/ng572. PMID 11479594.
- ^ Bei-sha, Tang et al (2005). "Novel compound heterozygous mutations in the PANK2 gene in a Chinese patient with atypical pantothenate kinase-associated neurodegeneration". Movement Disorders 20 (7): 819–21. doi:10.1002/mds.20408. PMC 2105744. PMID 15747360. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2105744.
- ^ http://www.britannica.com/bps/additionalcontent/18/27764296/Founder-mutation-in-the-PANK-gene-of-Agrawal-children-with-Neurodegeneration-with-Brain-Iron-accumulation-NBIA
- ^ Malmström-Groth AG, Kristensson K (1982). "Neuroaxonal dystrophy in childhood. Report of two second cousins with PKAN, and a case of Seitelberger's disease". Acta paediatrica Scandinavica 71 (6): 1045–9. doi:10.1111/j.1651-2227.1982.tb09574.x. PMID 7158329.
- ^ Jankovic J, Kirkpatrick JB, Blomquist KA, Langlais PJ, Bird ED (February 1985). "Late-onset Hallervorden-Spatz disease presenting as familial parkinsonism". Neurology 35 (2): 227–34. doi:10.1159/000153550. PMID 3969211.
- ^ Jankovic J, Kirkpatrick JB, Blomquist KA, Langlais PJ, Bird ED (1985). "Late-onset Hallervorden-Spatz disease presenting as familial parkinsonism". Neurology 35 (2): 227–34. doi:10.1159/000153550. PMID 3969211.
- ^ Tanfani G, Mascalchi M, Dal Pozzo GC, Taverni N, Saia A, Trevisan C (1987). "MR imaging in a case of Hallervorden-Spatz disease". Journal of computer assisted tomography 11 (6): 1057–8. doi:10.1097/00004728-198711000-00027. PMID 3680689.
- ^ Sethi KD, Adams RJ, Loring DW, el Gammal T (1988). "Hallervorden-Spatz syndrome: clinical and magnetic resonance imaging correlations". Ann. Neurol. 24 (5): 692–4. doi:10.1002/ana.410240519. PMID 3202617.
- ^ Angelini L, Nardocci N, Rumi V, Zorzi C, Strada L, Savoiardo M (1992). "Hallervorden-Spatz disease: clinical and MRI study of 11 cases diagnosed in life". J. Neurol. 239 (8): 417–25. doi:10.1007/BF00856805. PMID 1447570.
- ^ Casteels I, Spileers W, Swinnen T, et al. (1994). "Optic atrophy as the presenting sign in Hallervorden-Spatz syndrome". Neuropediatrics 25 (5): 265–7. doi:10.1055/s-2008-1073034. PMID 7885538.
- ^ Malandrini A, Bonuccelli U, Parrotta E, Ceravolo R, Berti G, Guazzi GC (1995). "Myopathic involvement in two cases of Hallervorden-Spatz disease". Brain Dev. 17 (4): 286–90. doi:10.1016/0387-7604(95)00039-E. PMID 7503394.
- ^ Taylor TD, Litt M, Kramer P, Pandolfo M, Angelini L, Nardocci N, Davis S, Pineda M, Hattori H, Flett PJ, Cilio MR, Bertini E, Hayflick SJ (1996). "Homozygosity mapping of Hallervorden-Spatz syndrome to chromosome 20p12.3-p13". Nat. Genet. 14 (4): 479–81. doi:10.1038/ng1296-479. PMID 8944032.
- ^ Pellecchia MT, Valente EM, Cif L, et al. (2005). "The diverse phenotype and genotype of pantothenate kinase-associated neurodegeneration". Neurology 64 (10): 1810–2. doi:10.1212/01.WNL.0000161843.52641.EC. PMID 15911822.
External links
Metabolic disorders of vitamins, coenzymes, and cofactors B7 Biotin/MCD Other B B5 (Pantothenate kinase-associated neurodegeneration) · B12 (Methylmalonic acidemia)Other vitamin Nonvitamin cofactor M: NUT
cof, enz, met
noco, nuvi, sysi/epon, met
drug(A8/11/12)
Categories:- Extrapyramidal and movement disorders
- Vitamin, coenzyme, and cofactor metabolism disorders
Wikimedia Foundation. 2010.