- Motor neurone disease
-
Motor neurone disease Classification and external resources 
spinal diagramICD-10 G12.2 ICD-9 335.2 DiseasesDB 8358 MeSH D016472 The motor neurone diseases (or motor neuron diseases) (MND) are a group of neurological disorders that selectively affect motor neurones,[1] the cells that control voluntary muscle activity including speaking, walking, breathing, swallowing and general movement of the body. They are generally progressive in nature, and can cause progressive disability and death. The drug riluzole can slow progression slightly. The condition was first described in full by the French neurologist Jean-Martin Charcot in 1869.
Contents
Classification
There are five recognized subtypes of motor neurone diseases. They are distinguished by the nerve cells affected (upper and lower motor neuron), and the symptoms that result from this damage. The "bulbar region" in the table below refers to the mouth, face, and throat.
Type UMN degeneration LMN degeneration ALS yes yes PLS yes no PMA no yes Progressive bulbar palsy no yes - bulbar region Pseudobulbar palsy yes - bulbar region no The disease spinal muscular atrophy (SMA) is classified under MND by the disease terminology classification system MeSH, but not in the tenth International Statistical Classification of Diseases and Related Health Problems published in 1992.
Signs and symptoms
Muscle symptoms
Symptoms usually present themselves between the ages of 50-70, and include progressive weakness, muscle wasting, and muscle fasciculations, spasticity or stiffness in the arms and legs, and overactive tendon reflexes. Patients may present with symptoms as diverse as a dragging foot, unilateral muscle wasting in the hands, or slurred speech.
Neurological examination presents specific signs associated with upper and lower motor neurone degeneration. Signs of upper motor neurone damage include spasticity, brisk reflexes and what are referred to as pathological reflexes (reflexes that may be normal in an infant but are abnormal in an adult) including Hoffmann's sign in the fingers and hand and the Babinski sign. Signs of lower motor neurone damage include weakness, fasciculations (twitching muscles) and muscle atrophy.
Every muscle group in the body requires both upper and lower motor neurones to function. The signs described above can occur in any muscle group, including the arms, legs, torso, and bulbar region.
The symptoms described above may resemble a number of other rare diseases, known as "MND Mimic Disorders". These include, but are not limited to, multifocal motor neuropathy, Kennedy's disease, hereditary spastic paraplegia, spinal muscular atrophy and monomelic amyotrophy. A small subset of familial MND cases occur in children, such as "juvenile ALS", Madras syndrome, and individuals who have inherited the ALS2 gene. However, these are not typically referred to as MND, but by their specific names.
Effects on cognition, mood, and senses
Around a third of all MND patients experience labile affect, also known as emotional lability, pseudobulbar affect, or pathological laughter and crying. Patients with pseudobulbar palsy are particularly likely to be affected, as are patients with PLS.
Cognitive change occurs in between 33–50% of patients. A small proportion exhibit a form of frontotemporal dementia characterised by behavioural abnormalities such as disinhibition, apathy, and personality changes. A small proportion of patients may also suffer from an aphasia, which causes difficulty in naming specific objects. A larger proportion (up to 50%) suffer from a milder version of cognitive change which primarily affects what is known as executive function. Briefly, this is the ability of an individual to initiate, inhibit, sustain, and switch attention and is involved in the organisation of complex tasks down to smaller components. Often patients with such changes find themselves unable to do the family finances or drive a car. Depression is surprisingly rare in MND (around 5–20%) relative to the frequency with which it is found in other, less severe, neurological disorders e.g. ~50% in multiple sclerosis and Parkinson's disease, ~20% in Epilepsy. Depression does not necessarily increase as the symptoms progress, and in fact many patients report being happy with their quality of life despite profound disability. This may reflect the use of coping strategies such as reevaluating what is important in life.
Although traditionally thought only to affect the motor system, sensory abnormalities are not necessarily absent, with some patients finding altered sensation to touch and heat, found in around 10% of patients. Patients with a predominantly upper motor neurone syndrome, and particularly PLS, often report an enhanced startle reflex to loud noises.
Neuroimaging and neuropathology has demonstrated extra-motor changes in the frontal lobes including the inferior frontal gyrus, superior frontal gyrus, anterior cingulate cortex, and superior temporal gyrus. The degree of pathology in these areas has been directly related to the degree of cognitive change experienced by the patient, if any. Patients with MND and dementia have been shown to exhibit marked frontotemporal lobe atrophy as revealed by MRI or SPECT neuroimaging.
Causes
About 90% of cases of MND are "sporadic", meaning that the patient has no family history of ALS and the case appears to have occurred with no known cause. Genetic factors are suspected to be important in determining an individual's susceptibility to disease, and there is some weak evidence to suggest that onset can be "triggered" by as yet unknown environmental factors (see 'Epidemiology' below).
Approximately 10% of cases are "familial MND", defined either by a family history of MND or by testing positive for a known genetic mutation associated with the disease. The following genes are known to be linked to ALS: Cu/Zn superoxide dismutase SOD1, ALS2, NEFH (a small number of cases), senataxin (SETX) and vesicle associated protein B (VAPB).
Of these, SOD1 mutations account for some 20% of familial MND cases. The SOD1 gene codes for the enzyme superoxide dismutase, a free radical scavenger that reduces the oxidative stress of cells throughout the body. So far over 100 different mutations in the SOD1 gene have been found, all of which cause some form of ALS(ALSOD database). In North America, the most commonly occurring mutation is known as A4V and occurs in up to 50% of SOD1 cases. In people of Scandinavian extraction there is a relatively benign mutation called D90A which is associated with a slow progression. In Japan, the H46R mutation is most common. G93A, the mutation used to generate the first animal model (and by far the most widely studied), is present only in a few families worldwide. Future research is concentrating on identifying new genetic mutations and the clinical syndrome associated with them. Familial MND may also confer a higher risk of developing cognitive changes such as frontotemporal dementia or executive dysfunction (see 'extra-motor change in MND' below).
It is thought that SOD1 mutations confer a toxic gain, rather than a loss, of function to the enzyme. SOD1 mutations may increase the propensity for the enzyme to form protein aggregates which are toxic to nerve cells.
Pathophysiology
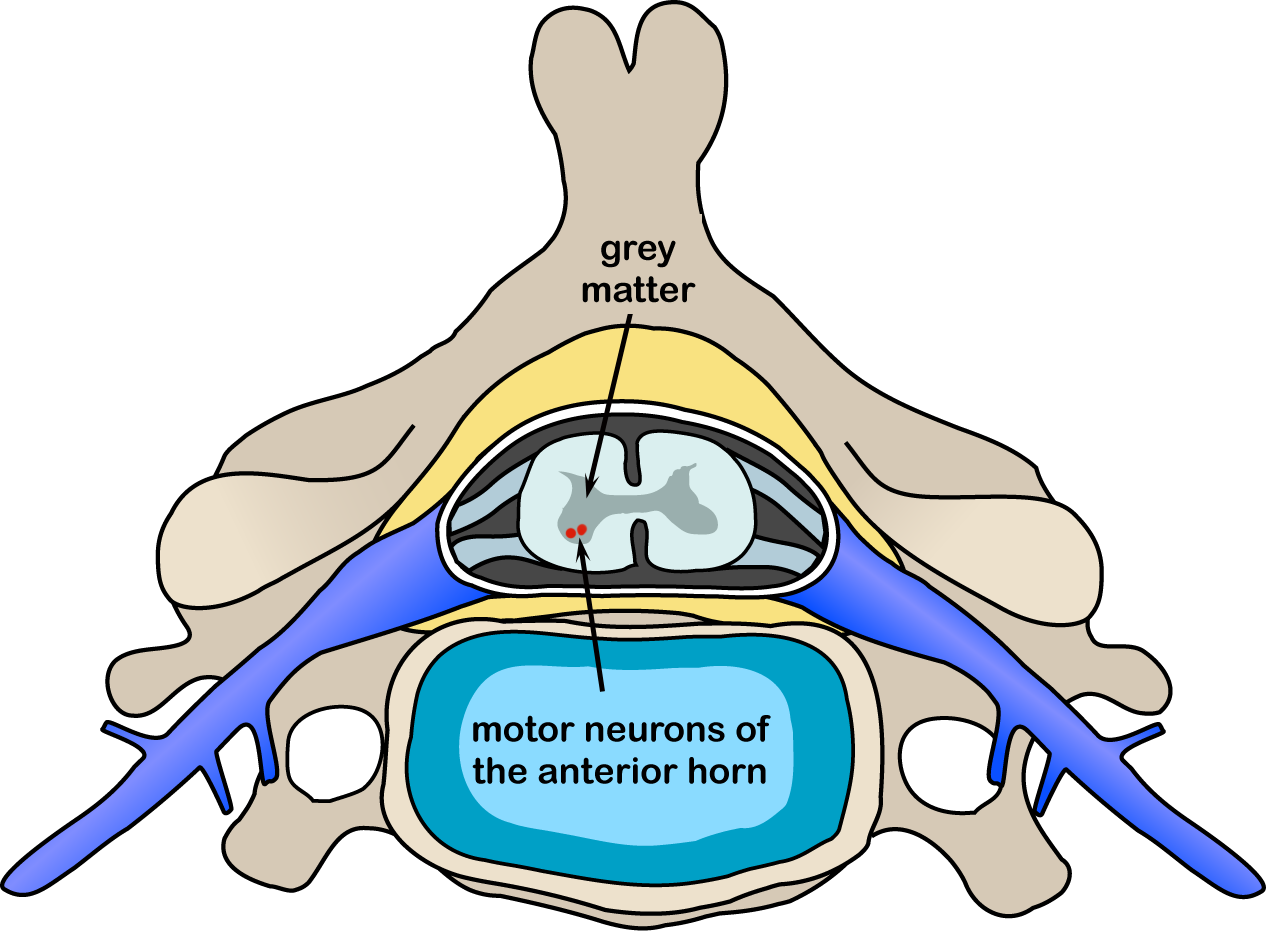
Skeletal muscles are innervated by a group of neurones (lower motor neurones) located in the ventral horns of the spinal cord which project out the ventral roots to the muscle cells. These nerve cells are themselves innervated by the corticospinal tract or upper motor neurones that project from the motor cortex of the brain. On macroscopic pathology, there is a degeneration of the ventral horns of the spinal cord, as well as atrophy of the ventral roots. In the brain, atrophy may be present in the frontal and temporal lobes. On microscopic examination, neurones may show spongiosis, the presence of astrocytes, and a number of inclusions including characteristic "skein-like" inclusions, Bunina bodies, and vacuolisation. Bunina bodies were first described in 1962; they are markers of neuronal degeneration.
The availability of mouse models has led to extensive research into the causes of SOD1-mutant linked familial ALS. The most commonly used mouse model is G93A,[2] although many others have since been generated. At the gross physiological level, the mouse models faithfully recapitulate the features of human ALS (motorneuron death, muscle atrophy, respiratory failure).
Although there is no consensus as to the exact mechanism by which mutated SOD1 causes the disease (in either mice or patients), studies based largely on mouse models suggest a role for excitotoxicity and more controversially, oxidative stress, presumably secondary to mitochondrial dysfunction. Death by apoptosis has also been suggested.
Diagnosis
The diagnosis of MND is a clinical one, established by a neurologist on the basis of history and neurological examination. There is no diagnostic test for MND. Investigations such as blood tests, electromyography (EMG), magnetic resonance imaging (MRI), and sometimes genetic testing are useful to rule out other disorders that may mimic MND. However, the diagnosis of MND remains a clinical one. Having excluded other diseases, a relatively rapid progression of symptoms is a strong diagnostic factor. Although an individual's progression may sometimes "plateau", it will not improve.
A set of diagnostic criteria called the El Escorial criteria[3][4] have been defined by the World Federation of Neurologists for use in research, particularly as inclusion/exclusion criteria for clinical trials. Owing to a lack of clinical diagnostic criteria, some neurologists use the El Escorial criteria during the diagnostic process, although strictly speaking this is functionality creep, and some have questioned the appropriateness of the criteria in a clinical setting.[5]
Treatment
Currently there is no cure for ALS. The only drug that affects the course of the disease is riluzole. The drug functions by blocking the effects of the neurotransmitter glutamate, and is thought to extend the lifespan of an ALS patient by only a few months.[6]
The lack of effective medications to slow the progression of ALS does not mean that patients with ALS cannot be medically cared for. Instead, treatment of patients with ALS focuses on the relief of symptoms associated with the disease. This involves a variety of health professionals including neurologists, speech-language pathologists, physical therapists, occupational therapists, dieticians, respiratory therapists, social workers, palliative care specialists, specialist nurses and psychologists.
Prognosis
Most cases of MND progress quite quickly, with noticeable decline occurring over the course of months. Although symptoms may present in one region, they will typically spread. If restricted to one side of the body they are more likely to progress to the same region on the other side of the body before progressing to a new region. After several years, most patients require help to carry out activities of daily living such as self care, feeding, and transportation.
MND is typically fatal within 2–5 years. Around 50% die within 14 months of diagnosis. The remaining 50% will not necessarily die within the next 14 months as the distribution is significantly skewed. As a rough estimate, 1 in 5 patients survive for 5 years, and 1 in 10 patients survive 10 years.[citation needed] Professor Stephen Hawking is a well-known example of a person with MND, and has lived for nearly 50 years with the disease. The television film Hawking starring Benedict Cumberbatch was made in 2004 about him and this disease.
Mortality normally results when control of the diaphragm is impaired and the ability to breathe is lost. One exception is Primary Lateral Sclerosis (PLS), which may last for upwards of 25 years. Given the typical age of onset, this effectively leaves most PLS patients with a normal life span. PLS can progress to ALS, decades later.
Epidemiology
The incidence of MND is approximately 1–5 out of 100,000 people. Men have a slightly higher incidence rate than women. Approximately 5,600 cases are diagnosed in the U.S. every year. By far the greatest risk factor is age, with symptoms typically presenting between the ages of 50-70. Cases under the age of 50 years are called "young onset MND", whilst incidence rates appear to tail off after the age of 85.
There are three "hot spots" of MND in the world. One is in the Kii peninsula of Japan, one amongst a tribal population in Papua New Guinea. Chamorro inhabitants from the island of Guam in the Pacific Ocean have an increased risk of developing a form of MND known as Guamanian ALS-PD-dementia complex or "lytico bodig", although the incidence rate has declined over the last 50 years and the average age of onset has increased.[7] Putative theories involve neurotoxins in the traditional diet including cycad nut flour and bats that have eaten cycad nuts.[8][9]
Etymology
Terminology regarding the motor neurone diseases can be confusing; in the UK "motor neurone disease" refers to both ALS specifically (the most common form of disease) and to the broader spectrum of motor neurone diseases including progressive muscular atrophy, primary lateral sclerosis, and progressive bulbar palsy. In the United States the most common terms used are ALS (both specifically for ALS and as a blanket term) or "Lou Gehrig's disease".[10] In France the disease is sometimes known as maladie de Charcot (Charcot's disease), although it may also be referred to by the direct translation of ALS, sclerose laterale amyotrophique (SLA). To avoid confusion, the annual scientific research conference dedicated to the study of MND is called the International ALS/MND Symposium. ALS/MND refers to a specific subset of pathologically identical diseases; there are numerous other afflictions of motor neurones that are pathologically distinct from ALS/MND and have a different clinical course. Examples of other diseases of the motor neurone that should not be confused with ALS/MND include spinobulbar muscular atrophy, spinal muscular atrophy, Charcot-Marie-Tooth disease, and many others.
Amyotrophic comes from the Greek language: A- means "no", myo refers to "muscle", and trophic means "nourishment"; amyotrophic therefore means "no muscle nourishment," which describes the characteristic atrophication of the sufferer's disused muscle tissue. Lateral identifies the areas in a person's spinal cord where portions of the nerve cells that are affected are located. As this area degenerates it leads to scarring or hardening ("sclerosis") in the region.
History
Although other 19th century neurologists previously described the disease, a French neurologist, Jean-Martin Charcot, first suggested grouping together disparate conditions that affect the lateral horn of the spinal cord in 1869.
See also
- Category:People with motor neurone disease
- Category:Deaths from motor neurone disease
- Kennedy's disease
- Monomelic amyotrophy
- Primary lateral sclerosis
- Progressive muscular atrophy
- Riluzole
References
- ^ "motor neuron disease" at Dorland's Medical Dictionary
- ^ Gurney, ME; Pu, H; Chiu, AY; Dal Canto, MC; Polchow, CY; Alexander, DD; Caliendo, J; Hentati, A et al. (1994). "Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation". Science 264 (5166): 1772–5. doi:10.1126/science.8209258. PMID 8209258.
- ^ Brooks BR (1994). "El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial "Clinical limits of amyotrophic lateral sclerosis" workshop contributors". J. Neurol. Sci. 124 Suppl: 96–107. doi:10.1016/0022-510X(94)90191-0. PMID 7807156.
- ^ "El Escorial Revisited: Revised Criteria for the Diagnosis of Amyotrophic Lateral Sclerosis - Requirements for Diagnosis". Archived from the original on 2007-05-17. http://web.archive.org/web/20070517041851/http://www.wfnals.org/guidelines/1998elescorial/elescorial1998criteria.htm. Retrieved 2007-06-06.
- ^ Belsh JM (2000). "ALS diagnostic criteria of El Escorial Revisited: do they meet the needs of clinicians as well as researchers?". Amyotroph. Lateral Scler. Other Motor Neuron Disord. 1 Suppl 1: S57–60. doi:10.1080/14660820052415925. PMID 11464928.
- ^ Miller, R. G.; Mitchell, J.; Lyon, M.; Moore, D. (2007). "Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND)". Cochrane Database of Systematic Reviews (1): CD001447. doi:10.1002/14651858.CD001447.pub2. PMID 17253460.
- ^ Waring, S.C et al. (2004). "Incidence of Amyotrophic Lateral Sclerosis and of the Parkinsonism-Dementia Complex of Guam, 1950-1989". Neuroepidemiology 23 (4): 192–199. doi:10.1159/000078505. PMID 15272222.
- ^ "Flying fox linked to disease - The Boston Globe". 2003-12-09. http://www.boston.com/news/science/articles/2003/12/09/flying_fox_linked_to_disease/. Retrieved 2007-06-06.
- ^ Miller G (2006). "Neurodegenerative disease. Guam's deadly stalker: on the loose worldwide?". Science 313 (5786): 428–31. doi:10.1126/science.313.5786.428. PMID 16873621. http://www.sciencemag.org/cgi/content/full/313/5786/428.
- ^ "Motor Neuron Diseases Fact Sheet: National Institute of Neurological Disorders and Stroke (NINDS)". www.ninds.nih.gov. http://www.ninds.nih.gov/disorders/motor_neuron_diseases/detail_motor_neuron_diseases.htm. Retrieved 2010-11-07.
External links
- MND Association
- MND Scotland
- motor_neuron_diseases at NINDS
- MND Association of Australia
- Motor Neuron Disease Association of South Africa
- ALS Association (US-based)
- ALS Society of Canada
- Project A.L.S
- ALSOD Database of all known SOD1 mutations
- ALS Therapy Development Foundation
Categories:- Disability
- Motor neurone disease
- Systemic atrophies primarily affecting the central nervous system
- Rare diseases
Wikimedia Foundation. 2010.