- Maple syrup urine disease
-
Maple Syrup Urine Disease Classification and external resources 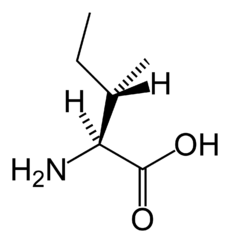
Isoleucine (pictured above), leucine, and valine are the branched-chain amino acids that build up in MSUD.ICD-10 E71.0 ICD-9 270.3 OMIM 248600 DiseasesDB 7820 MedlinePlus 000373 eMedicine ped/1368 MeSH D008375 Maple syrup urine disease (MSUD), also called branched-chain ketoaciduria, is an autosomal recessive[1] metabolic disorder affecting branched-chain amino acids. It is one type of organic acidemia.[2] The condition gets its name from the distinctive sweet odor of affected infants' urine.[3]
Contents
Diagnosis and symptoms
MSUD is caused by a deficiency of the branched-chain alpha-keto acid dehydrogenase complex (BCKDC), leading to a buildup of the branched-chain amino acids (leucine, isoleucine, and valine) and their toxic by-products in the blood and urine.
The disease is characterized in an infant by the presence of sweet-smelling urine, with an odor similar to that of maple syrup. Infants with this disease seem healthy at birth but if left untreated suffer severe brain damage and eventually die.
From early infancy, symptoms of the condition include poor feeding, vomiting, dehydration, lethargy, hypotonia, seizures, hypoglycaemia, ketoacidosis, opisthotonus, pancreatitis, coma and neurological decline.
Classification
Maple syrup urine disease can be classified by its pattern of signs and symptoms, or by its genetic cause. The most common and severe form of the disease is the classic type, which appears soon after birth. Variant forms of the disorder may appear later in infancy or childhood and are typically less severe, but still involve mental and physical problems if left untreated.
There are several variations of the disease:
- Classic Severe MSUD
- Intermediate MSUD
- Intermittent MSUD
- Thiamine-responsive MSUD
- E3-Deficient MSUD with Lactic Acidosis
Management
Keeping MSUD under control requires careful monitoring of blood chemistry and involves both a special diet and frequent testing.
A diet with minimal levels of the amino acids leucine, isoleucine, and valine must be maintained in order to prevent neurological damage. As these three amino acids are required for proper metabolic function in all people, specialized protein preparations containing substitutes and adjusted levels of the amino acids have been synthesized and tested, allowing MSUD patients to meet normal nutritional requirements without causing harm.[4]
Usually, patients are also monitored by a dietitian. Their diet must be adhered to strictly and permanently. However, with proper management, those afflicted are able to live healthy, normal lives and not suffer the severe neurological damage associated with the disease.
Genetic prevalence
 Maple syrup urine disease has an autosomal recessive pattern of inheritance.
Maple syrup urine disease has an autosomal recessive pattern of inheritance.
Maple syrup urine disease affects approximately 1 out of 180,000 infants.[5] Due in part to the founder effect,[6] however, MSUD has a much higher prevalence in children of Amish, Mennonite, and Jewish descent.[7][5][8]
Mutations in the following genes cause maple syrup urine disease:
- BCKDHA (Online 'Mendelian Inheritance in Man' (OMIM) 608348)
- BCKDHB (Online 'Mendelian Inheritance in Man' (OMIM) 248611)
- DBT (Online 'Mendelian Inheritance in Man' (OMIM) 248610)
- DLD (Online 'Mendelian Inheritance in Man' (OMIM) 238331)
These four genes produce proteins that work together as the branched-chain alpha-keto acid dehydrogenase complex. The complex is essential for breaking down the amino acids leucine, isoleucine, and valine, which are present in many kinds of food (in particular, protein-rich foods such as milk, meat, and eggs). Mutations in any of these genes reduce or eliminate the function of the enzyme complex, preventing the normal breakdown of isoleucine, leucine, and valine. As a result, these amino acids and their by-products build up in the body. Because high levels of these substances are toxic to the brain and other organs, this accumulation leads to the serious medical problems associated with maple syrup urine disease.
This condition has an autosomal recessive inheritance pattern, which means the defective gene is located on an autosome, and two copies of the gene - one from each parent - must be inherited to be affected by the disorder. The parents of a child with an autosomal recessive disorder are carriers of one copy of the defective gene, but are usually not affected by the disorder.
See also
References
- ^ Podebrad F, Heil M, Reichert S, Mosandl A, Sewell AC, Böhles H (April 1999). "4,5-dimethyl-3-hydroxy-25H-furanone (sotolone)--the odour of maple syrup urine disease". Journal of inherited metabolic disease 22 (2): 107–114. doi:10.1023/A:1005433516026. PMID 10234605.
- ^ Ogier de Baulny H, Saudubray JM (2002). "Branched-chain organic acidurias". Semin Neonatol. 7 (1): 65–74. doi:10.1053/siny.2001.0087. PMID 12069539.
- ^ [1]
Genetics Home Reference (GRH) - Genetic Conditions - Maple Syrup Urine Disease. GHR is a service of the U.S. National Library of Medicine. Reviewed February 2006; Published June 12, 2009 - ^ Hallam P, Lilburn M, Lee PJ (2005). "A new protein substitute for adolescents and adults with maple syrup urine disease (MSUD)". J. Inherit. Metab. Dis. 28 (5): 665–672. doi:10.1007/s10545-005-0061-6. PMID 16151896.
- ^ a b http://rarediseases.about.com/od/rarediseases1/a/062004.htm
About.com: Rare diseases - June 2004. Maple syrup urine disease by Mary Kugler, R.N.
Article describes MSUD prevalence among Amish and Mennonite children. - ^ Jaworski MA, Severini A, Mansour G, Konrad HM, Slater J, Henning K, Schlaut J, Yoon JW, Pak CY, Maclaren N, et al. (1989). "Genetic conditions among Canadian Mennonites: evidence for a founder effect among the old country (Chortitza) Mennonites". Clin Invest Med 12 (2): 127–141. PMID 2706837.
- ^ Puffenberger EG (2003). "Genetic heritage of Old Order Mennonites in southeastern Pennsylvania". Am J Med Genet C Semin Med Genet. 121 (1): 18–31. doi:10.1002/ajmg.c.20003. PMID 12888983.
- ^ http://www.mazornet.com/genetics/MapleSyrup.htm
External links
- Maple syrup urine disease at NLM Genetics Home Reference
- Maple syrup urine disease at GPnotebook
- msud at NIH/UW GeneTests
- Maple Syrup Urine Disease (Overview on NLM)
- Maple syrup urine disease at the Open Directory Project
Inborn error of amino acid metabolism (E70–E72, 270) K→acetyl-CoA Lysine/straight chainMaple syrup urine disease · Isovaleric acidemia · 3-Methylcrotonyl-CoA carboxylase deficiency · 3-hydroxy-3-methylglutaryl-CoA lyase deficiency · 3-Methylglutaconic aciduria 1HypertryptophanemiaG G→pyruvate→citrateG→glutamate→
α-ketoglutarateGlutamate/glutamineMaple syrup urine disease · Beta-ketothiolase deficiency · 2-Methylbutyryl-CoA dehydrogenase deficiencyG→fumarateType II tyrosinemia · Type III tyrosinemia/Hawkinsinuria · Alkaptonuria/Ochronosis · Type I tyrosinemiaTransport/
IE of RTTOther Categories:- Amino acid metabolism disorders
- Rare diseases
- Autosomal recessive disorders

Wikimedia Foundation. 2010.