- Ornithine translocase deficiency
-
Ornithine translocase deficiency Classification and external resources 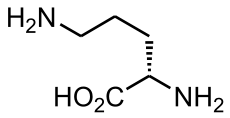
OrnithineICD-9 270.6 OMIM 238970 DiseasesDB 29849 Ornithine translocase deficiency, also called Hyperornithinemia-Hyperammonemia-Homocitrullinuria (HHH) syndrome,[1] is a rare autosomal recessive[2] urea cycle disorder affecting the enzyme ornithine translocase, which causes ammonia to accumulate in the blood, a condition called hyperammonemia.
Ammonia, which is formed when proteins are broken down in the body, is toxic if the levels become too high. The nervous system is especially sensitive to the effects of excess ammonia.
Contents
Diagnosis
Ornithine translocase deficiency varies widely in its severity and age of onset. An infant with ornithine translocase deficiency may be lacking in energy (lethargic) or refuse to eat, or have poorly controlled breathing or body temperature. Some babies with this disorder may experience seizures or unusual body movements, or go into a coma. Episodes of illness may coincide with the introduction of high-protein formulas or solid foods into the diet.
In most affected individuals, signs and symptoms of ornithine translocase deficiency do not appear until later in life. Later-onset forms of ornithine translocase deficiency are usually less severe than the infantile form. Some people with later-onset ornithine translocase deficiency cannot tolerate high-protein foods, such as meat. Occasionally, high-protein meals or stress caused by illness or periods without food (fasting) may cause ammonia to accumulate more quickly in the blood. This rapid increase of ammonia may lead to episodes of vomiting, lack of energy (lethargy), problems with coordination (ataxia), confusion, or blurred vision. Complications of ornithine translocase deficiency may include developmental delay, learning disabilities, and stiffness caused by abnormal tensing of the muscles (spasticity).
Pathophysiology
 Ornithine translocase deficiency has an autosomal recessive pattern of inheritance.
Ornithine translocase deficiency has an autosomal recessive pattern of inheritance.
Mutations in the SLC25A15 gene cause ornithine translocase deficiency. Ornithine translocase deficiency belongs to a class of metabolic disorders referred to as urea cycle disorders. The urea cycle is a sequence of reactions that occurs in liver cells. This cycle processes excess nitrogen, generated when protein is used by the body, to make a compound called urea that is excreted by the kidneys. The SLC25A15 gene provides instructions for making a protein called a mitochondrial ornithine transporter. This protein is needed to move a molecule called ornithine within the mitochondria (the energy-producing centers in cells). Specifically, this protein transports ornithine across the inner membrane of mitochondria to the region called the mitochondrial matrix, where it participates in the urea cycle. Mutations in the SLC25A15 gene result in a mitochondrial ornithine transporter that is unstable or the wrong shape, and which cannot bring ornithine to the mitochondrial matrix. This failure of ornithine transport causes an interruption of the urea cycle and the accumulation of ammonia, resulting in the signs and symptoms of ornithine translocase deficiency.
This disorder is inherited in an autosomal recessive pattern, which means the defective gene is located on an autosome, and two copies of the gene - one from each parent - are required to be born with the disorder. The parents of an individual with an autosomal recessive disorder each carry one copy of the altered gene but do not show signs and symptoms of the disorder.
See also
- Ornithine transcarbamylase deficiency
- Inborn errors of metabolism
- Gyrate atrophy
External links
- Ornithine translocase deficiency at NLM Genetics Home Reference
- For a thorough scientific overview of hyperornithinemias, one can consult chapter 83 of The Online Metabolic and Molecular Bases of Inherited Disease.[3]
References
- ^ Online 'Mendelian Inheritance in Man' (OMIM) 238970
- ^ Hommes FA, Roesel RA, Metoki K, Hartlage PL, Dyken PR (Feb 1986). "Studies on a case of HHH-syndrome (hyperammonemia, hyperornithinemia, homocitrullinuria)". Neuropediatrics 17 (1): 48–52. doi:10.1055/s-2008-1052499. ISSN 0174-304X. PMID 3960284.
- ^ Charles Scriver, Beaudet, A.L., Valle, D., Sly, W.S., Vogelstein, B., Childs, B., Kinzler, K.W. (Accessed 2007). New York: McGraw-Hill. - Summaries of 255 chapters, full text through many universities. There is also the OMMBID blog.
Inborn error of amino acid metabolism (E70–E72, 270) K→acetyl-CoA Lysine/straight chainHypertryptophanemiaG G→pyruvate→citrateG→glutamate→
α-ketoglutarateGlutamate/glutamineG→fumarateType II tyrosinemia · Type III tyrosinemia/Hawkinsinuria · Alkaptonuria/Ochronosis · Type I tyrosinemiaTransport/
IE of RTTOther Categories:- Amino acid metabolism disorders
- Autosomal recessive disorders
- Rare diseases


Wikimedia Foundation. 2010.