- Cystathioninuria
-
Cystathioninuria Classification and external resources 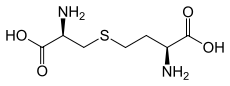
CystathionineICD-10 E72.1 ICD-9 270.4 OMIM 219500 DiseasesDB 29671 Cystathioninuria, also called cystathionase deficiency, is an autosomal recessive[1] metabolic disorder that results in an excess of cystathionine in the urine. It is associated with a congenital dysfunction of the enzyme cystathionase, or acquired deficiency of vitamin B6 which is essential for the function of this enzyme. The latter is usually related to an overall deficiency of all the B-complex vitamins.
Genetics
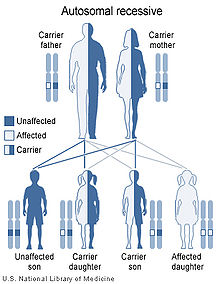 Cystathioninuria has an autosomal recessive pattern of inheritance.
Cystathioninuria has an autosomal recessive pattern of inheritance.
Cystathioninuria is inherited in an autosomal recessive manner.[1] This means the defective gene responsible for the disorder is located on an autosome, and two copies of the defective gene (one inherited from each parent) are required in order to be born with the disorder. The parents of an individual with an autosomal recessive disorder both carry one copy of the defective gene, but usually do not experience any signs or symptoms of the disorder.
Pathophysiology
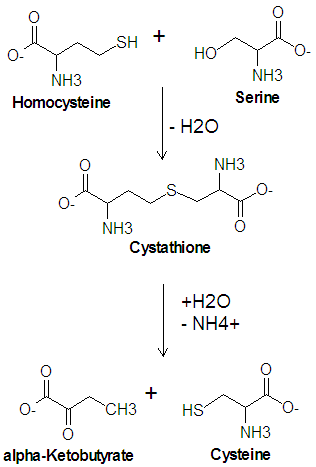
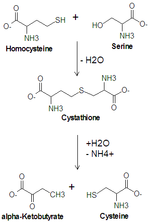 Cysteine metabolism. Cystathionine gamma-lyase (cystathionase) catalyzes the lower reaction.
Cysteine metabolism. Cystathionine gamma-lyase (cystathionase) catalyzes the lower reaction.References
- ^ a b Online 'Mendelian Inheritance in Man' (OMIM) 219500
Inborn error of amino acid metabolism (E70–E72, 270) K→acetyl-CoA Lysine/straight chainHypertryptophanemiaG G→pyruvate→citrateG→glutamate→
α-ketoglutarateGlutamate/glutamineG→fumarateType II tyrosinemia · Type III tyrosinemia/Hawkinsinuria · Alkaptonuria/Ochronosis · Type I tyrosinemiaTransport/
IE of RTTOther Categories:- Amino acid metabolism disorders
- Autosomal recessive disorders
- Genetic disorder stubs

Wikimedia Foundation. 2010.